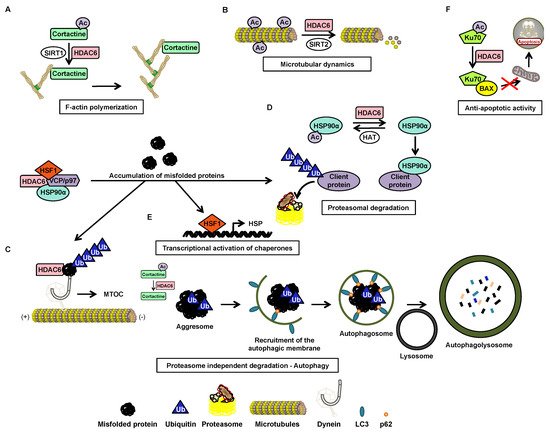
Histone deacetylase (HDAC) 6 is a zinc-dependent enzyme of HDAC class IIb. HDAC6 is unique within the HDAC family due to a particular structure giving it unique biological functions implicated in all major cell pathways. This isoenzyme is mainly active in the cytoplasm and possesses two functional catalytic sites and an ubiquitin-binding domain. The deacetylase functions of HDAC6 targets multiple substrates including essentially α-tubulin and heat shock protein (HSP)90α which are key factors in cell regulatory networks through the regulation of the microtubule network and many protein functions, respectively. Accordingly, several studies have highlighted the role of HDAC6 in various pathological conditions. For instance HDAC6 overexpression frequently correlates with tumorigenesis and favor cell survival and metastasis. Therefore, HDAC6 represents an interesting potential therapeutic target.
- histone deacetylase 6 inhibitor
- personalized treatment
- heat shock protein 90α
- leukemia stem cells
- imatinib resistance
- targeted therapy
1. Introduction
Carcinogenesis is a multistep process whereby normal cells are transformed into malignant cells. The process is characterized by major biological changes shared by most neoplastic cells called hallmarks of cancer. These transformational events relies on multiple alterations at genetic and epigenetic levels leading to abnormal cell growth [1].
Over the past years protein lysine acetylation has emerged as a key post-translational modification in the coordination of tightly regulated biological functions and alterations of the acetylome profiles are associated with various pathological conditions such as cancer.
The acetylation status of lysine residues within histone and non-histone proteins is finely tuned by the concert action of histone acetyltransferases (HATs) and histone deacetylases (HDACs) catalyzing the addition and removal of the acetyl groups, respectively. Recently, there was a particular focus on HDAC6 coming from its unique properties to control multiple cellular pathways linked to cell growth, survival, and migration. Accordingly, the use of HDAC6 inhibitors alone or in combination with additional chemotherapeutic agents appear as a promising strategy to treat various cancers.
2. Histone Deacetylase 6
2.1. Structure

2.2. Function
| Substrates | Localization of the Substrate | Deacetylated Lysine(s) | Function of the Deacetylated Substrate | Interaction Domains of HDAC6 | Reference |
|---|---|---|---|---|---|
| 14-3-3ζ | Cytoplasm and nucleus | 49, 120 | Regulation of protein binding Bad and AS160 | ND | [11] |
| β-catenin | Cytoplasm and nucleus | 49 | Epidermal growth factor-induced nuclear localization and decreased expression of c-Myc | ND | [9] |
| Cortactin * | Cytoplasm | 87, 124, 161, 189, 198, 235, 272, 309, 319 | Regulation of cell migration and actin filament binding | DD1 and DD2 | [9] |
| DNAJA1 | Cytoplasm | ND | Protein folding | ND | [12] |
| ERK1 | Cytoplasm and nucleus | 72 | Proliferation, mobility, and cell survival | ND | [13] |
| Foxp3 * | Nucleus | ND | ND | ND | [14] |
| HDAC9 | Cytoplasm and nucleus | ND | Modulation of cell survival and arrest of cellular movement | DD2 | [15] |
| HDAC11 | Nucleus | ND | Transcriptional activation of interleukin 10 | ND | [16] |
| HMGN2 | Nucleus | 2 | Increased transcription of STAT5 | ND | [17] |
| HSC70 | Cytoplasm | ND | Protein folding | ND | [12] |
| HSPA5 | Cytoplasm | 353 | Ubiquitination of HSPA5 mediated by GP78 | ND | [18] |
| HSP90α | Cytoplasm | 294 | Degradation and elimination of misfolded proteins and regulation of glucocorticoid receptors | DD1, DD2 et BUZ | [9] |
| K-RAS * | Cytoplasm | 104 | Cell proliferation | ND | [19] |
| Ku70 | Cytoplasm | 539, 542 | Suppression of apoptosis | ND | [9] |
| LC3B-II* | Cytoplasm | ND | Regulation of autophagy | ND | [20] |
| MSH2 | Cytoplasm and nucleus | 845, 847, 871, 892 | Reduced cellular sensitivity to DNA damaging agents and reduced DNA mismatch repair activities by downregulation of MSH2 | DD1 | [21] |
| MYH9 | Cytoplasm | ND | Regulation of binding to actin filaments | ND | [12] |
| PrxI | Cytoplasm and nucleus | 197 | Antioxidant activity | ND | [22][23] |
| PrxII | Cytoplasm and nucleus | 196 | Antioxidant activity | ND | [22][23] |
| RIG-I | Cytoplasm | 858, 909 | Recognition of viral RNA | ND | [24] |
| Sam68 | Nucleus | ND | Alternative splicing | ND | [25] |
| Survivin | Nucleus | 129 | Anti-apoptotic function | DD2 | [9] |
| Tat | Cytoplasm | 28 | Suppression of HIV transactivation | DD2 and BUZ | [26] |
| α-tubulin * | Cytoplasm | 40 | Formation of immune synapses, viral infection, cell migration and chemotaxis | DD1 or DD2 | [9][27] |
2.3. Post-Transcriptional Regulation
2.4. Post-Translational Regulation
| Post-Translational Modification | Enzyme | Target Site | Consequences | Reference |
|---|---|---|---|---|
| Phosphorylation | GSK3β | Ser-22 | Increased deacetylation activity of α-tubulin | [10] |
| ERK1 | Ser-1035 | Regulation of cellular motility | [10] | |
| GRK2 | ND | Increased deacetylation activity of α-tubulin | [32] | |
| Aurora | ND | Increased deacetylation activity of α-tubulin | [10] | |
| PKCζ | ND | Increased deacetylation activity of α-tubulin | [10] | |
| CK2 | Ser-458 | Improved formation and elimination of aggresomes | [10] | |
| EGFR | Tyr-570 | Inhibition of deacetylation activity | [33] | |
| Acetylation | p300 | Lys-16 | Inhibition of deacetylation activity | [10] |
| Protein Inhibiting HDAC6 by Direct Interaction | Protein Function | Protein Region Required for Interaction with HDAC6 | HDAC6 Domain Interacting with the Protein | Cellular Impact | References |
|---|---|---|---|---|---|
| CYLD | Deubiquitinase | ND | DD1/DD2 | Cell proliferation, ciliogenesis | [10] |
| Dysferlin | Skeletal muscle membrane repair, myogenesis, cell adhesion, intercellular calcium signaling | Domain C2 | ND | Myogenesis | [34] |
| Mdp3 | Stabilization factor of microtubules | Amino-terminal region | ND | Cell motility | [35] |
| Paxillin | Focal adhesion | Region rich in proline | ND | Polarization and cell migration | [10] |
| p62 | Transport of misfolded proteins | Between the ZZ domain and the TRAF6 link area | DD2 | Aggresome formation | [36] |
| RanBPM | Apoptosis, proliferation and cell migration | ND | Aggresome formation | [37] | |
| Tau | Stabilization factor of microtubules | Tubulin binding region | SE14 domain | Aggresome formation | [36][38] |
| TPPP1 | Polymerization and acetylation of microtubules | ND | Regulation of microtubule acetylation and β-catenin expression | [39] |
3. HDAC6 in cancer
Several studies have demonstrated the influence of HDAC6 in neurodegenerative, cardiovascular and renal diseases, as well as in inflammation [40] and viral response [10]. The role of the HDAC6 protein in cancer is also now well better understood. Although its oncogenic or tumor suppressor potential is dependent on the type of cancer [28], its involvement in oncogenic cell transformation, tumor development, and cancer immunity regulation makes a strong therapeutic candidate [41].
| Cancer Type | Cancers | Expression of HDAC6-Comments | References |
|---|---|---|---|
| Solid tumors | Bladder | Overexpressed | [41] |
| Melanoma | Overexpressed | [41] | |
| Lung | Overexpressed | [41] | |
| Oral squamous cell carcinoma | Overexpressed-Enhanced expression in advanced stages | [28][42] | |
| Ovarian carcinoma | Overexpressed-Enhanced expression in advanced stages | [28][42] | |
| Breast | Overexpressed-Prediction of a good or bad prognosis | [28][43] | |
| Hepatocytic carcinoma | Overexpressed-Enhanced expression in advanced stages | [28] | |
| Under-expressed-HDAC6 suggested as a tumor suppressor | [28][44] | ||
| Hematological | Chronic lymphocytic leukemia | Overexpressed-Observation on patient samples, cell lines and a transgenic mouse model | [42] |
| Acute myeloid leukemia | Overexpressed | [28][42] | |
| Acute lymphoblastic leukemia | Overexpressed-Enhanced expression in advanced stages | [28] | |
| Chronic lymphocytic leukemia | Overexpressed-Correlated with longer survival | [28] | |
| T-cell cutaneous lymphoma | Overexpressed-Correlated with longer survival | [28] | |
| Chronic myeloid leukemia | Overexpressed-Increased expression in CD34+ cells | [45] | |
| Multiple myeloma | Overexpressed | [46] | |
| Mantle cell lymphoma | Overexpressed | [46] | |
| Diffuse large B cell lymphoma | Overexpressed | [46] | |
| Peripheral T-cell lymphoma | Overexpressed | [46] |
3.1. HDAC6 Inhibitors
| Class | HDAC6 Inhibitor | Binding Domain | CI50 (nM) of the HDAC6 Activity in Vitro | Selectivity Ratio for HDAC6 Compared to (Other HDACs) | Inhibition of HDAC6 in Cellulo (µM)$ | Effect on Cancer Cell Lines or Cancer Type | References |
|---|---|---|---|---|---|---|---|
| Benzamides | Trithiocarbonate derivative (12ac) | ND | 65 | 19 (HDAC1) | 10 (lung cancer) | CI50 = 8.2 µM (cervical cancer) | [47] |
| NQN-1 (2-benzyl-amino-naphthoquinone) | ND | 5540 | Values non available (HDAC1, 2, 3, 4, 5, 7, 8, 9, 10, 11) | 4 (chronic myeloid leukemia) | CI50 = 0.8 µM (leukemia) | [48] | |
| Hydroxamates | Hydroxamic acid containing a phenylalanine (4n) | His215, His216, Tyr386, Phe283, and Tyr255 of DD1 and His610, His611, Tyr782, Phe620, and Phe680 of an HDAC6 homology model | 1690 | 14 (HDAC1) | 1 (colorectal carcinoma) | IC50: 3 to > 50 µM (various cancer cell lines) | [49] |
| Hydroxamic acid containing a pyridylalanine (5a) | Phe566 of DD2 of an HDAC6 homology model | 3970 | 25 (HDAC1) | ND | IC50: 104 µM (breast cancer) | [50] | |
| ACY-738 | ND | 1.7 | 55 (HDAC1), 75 (HDAC2), 128 (HDAC3) | 2.5 (neural cells) | ND | [51] | |
| ACY-775 | ND | 7.5 | 283 (HDAC1), 343 (HDAC2), 1496 (HDAC3) | 2.5 (neural cells) | ND | [51] | |
| ACY-1083 | His573 and His574 of DD2 | 3 | 260 (HDAC1) | 0.03 (neuroblastoma) | ND | [52][53] | |
| Bavarostat | Ser568 of DD2 | 60 | >10000 (HDAC1, 2, 3), 188 (HDAC4), 317 (HDAC5), 78 (HDAC7), 142 (HDAC8), 87 (HDAC9), >17 (HDAC10), 167 (HDAC11) | 10 (neural progenitor cells derived from induced pluripotent stem cells) | ND | [54] | |
| BRD9757 | ND | 30 | 21 (HDAC1), 60 (HDAC2), 23 (HDAC3), 727 (HDAC4), 611 (HDAC5), 420 (HDAC7), 36 (HDAC8), >1000 (HDAC9) | 10 (cervical cancer) | ND | [55] | |
| Cay10603 | His499 of DD2 of an HDAC6 homology model | 0.002 | ND | <1 to 1 µM (several pancreatic cancer cell lines) | ND | [56][57] | |
| Citarinostat (ACY-241) | ND | 2.6 | 14 (HDAC1), 17 (HDAC2), 18 (HDAC3 and 4), >7000 (HDAC4, 5,9), 2808 (HDAC7), 53 (HDAC8), | 0.3 (ovarian cancer) | CI50: 4.6 to 6.1 µM (ovarian and breast cancer) | [58] | |
| α3β-cyclic tetrapeptide (23) | ND | 39 | 3 (HDAC1), 4 (HDAC3), 6 (HDAC8) | 2 (acute lymphoblastic leukemia) | IC50: 9 to > 20 µM (various cancer cell lines) | [59] | |
| Compound containing a phenylisoxazole group as a surface recognition group (7) | His499 of HDAC7 | 0.002 | >100000 (HDAC1), >100000 (HDAC2), 210 (HDAC3), >3000000 (HDAC8), 45350 (HDAC10) | ND | IC50: 0.1 to 1 µM (various prostate cancer cell lines) | [56] | |
| Compound containing a triazolylphenyl group (6b) | ND | 1.9 | 52 (HDAC1), 155 (HDAC2), 7 (HDAC3), 420 (HDAC8), 59 (HDAC10) | ND | IC50: <0.5 to 22 µM (several prostate cancer lines) | [60] | |
| Compound containing a peptoid (2i) | Tyr301 of DD2 of an HDAC6 homology model | 1.59 | 126 (HDAC2), >6000 (HDAC4), 40 (HDAC11) | N | IC50: 0.34 to 2.7 µM (various cancer cell lines) | [61] | |
| 3-aminopyrrolidinone derivative (33) | ND | 17 | 4359 (HDAC1), 11 (HDAC8) | 0.3 (multiple myeloma) | Good oral bioavailability | [62] | |
| 4-aminomethylaryl acid derivative (1a) | ND | 19 | 305 (HDAC1), 842 (HDAC2), 237 (HDAC3), 790 (HDAC4), 174 (HDAC5), 242 (HDAC7), 36 (HDAC8), 195 (HDAC0) | 0.46 (cervical cancer) | ND | [63] | |
| 4-hydroxybenzoic acid derivative (7b) | ND | 200 | >50000 (HDAC1, 2, 8), >500000 (HDAC3, 10, 11) | 50 (prostate cancer) | IC50: 41 to 130 (several prostate and breast cancer cell lines) | [64] | |
| 4-hydroxybenzoic acid derivative (13a) | ND | 20000 | 25 (HDAC1), >5000 (HDAC2, 3, 4, 8, 10), >2500 (HDAC11) | 50 (prostate cancer) | IC50: 19 to 127 (several prostate and breast cancer cell lines) | [64] | |
| Aminoteraline derivative (32) | Phe620 and Phe680 of an HDAC6 homology model | 50 | 126 (HDAC1), 2 (HDAC8) | 2 (neuroblastoma) | IC50 = 5.4 µM (neuroblastoma) | [65] | |
| Benzothiophene derivative (39) | ND | 14 | ND | Same effect as tubastatin A | Does not target NF-κB and AP-1 at the transcriptional level | [66] | |
| 2,4-imidazolinedione derivative (10c) | ND | 4.4 | 218 (HDAC1), 63 (HDAC2), 53 (HDAC3), > 20000 (HDAC4, 7, 8, 9, 11), 3386 (HDAC5), 37 (HDAC10) | 1.6 (acute myeloid leukemia) | IC50: 0.2 to 0.8 µM (various cancer cell lines) | [67] | |
| Mercaptoacetamide derivative (2) | ND | 95.3 | 34 (HDAC1), 77 (HDAC2), 64 (HDAC8), 112 (HDAC10) | ND | At 10 µM protects cortical neurons from oxidative stress inducing death | [68] | |
| N-Hydroxycarbonylbenylamino quinoline derivative (13) | ND | 0.291 | 32817 (HDAC1), 42955 (HDAC2), 26632 (HDAC3), 15250 (HDAC4), 10694 (HDAC5), 2436 (HDAC7), 4089 (HDAC8), 5258 (HDAC9), 33646 (HDAC10), 1292 (HDAC11) | 0.1 (multiple myeloma) | IC50: 9.1 to 40.6 µM (multiple myeloma) | [69] | |
| Isoxazole-3-hydroxamate derivative (SS-208) | His463, Pro464, Phe583, and Leu712 of DD2 | 12 | 116 (HDAC1), 1625 (HDAC4), 576 (HDAC5), 695 (HDAC7), 103 (HDAC8), 3183 (HDAC9), 427 (HDAC11) | 5 (melanoma) | ND | [70] | |
| Phenothiazine derivative (7i) | Phe620 and Phe680 of DD2 | 5 | 538 (HDAC1) | 0.1 (acute myeloid leukemia) | ND | [71] | |
| Phenylhydroxamate derivative (2) | Phe464 and His614 of DD2 | 3 | 27 (HDAC1) | ND | CI50: 0.65 to 2.77 (ovarian cancer and squamous cell carcinoma of the tongue) | [61][72] | |
| Phenylsulfonylfuroxan derivative (5c) | ND | 7.4 | 33 (HDAC1), 51 (HDAC2), 45 (HDAC3), 4 (HDAC4), 46 (HDAC8), 82 (HDAC11) | 0.013 (acute myeloid leukemia) | IC50: 0.4 to 5.8 µM (various cancer cell lines) | [73] | |
| Pyridone derivative (11e) | Phe155 and Phe210 of HDAC2 | 2.46 | 8 (HDAC1), 52 (HDAC2), 127 (HDAC3), 2329 (HDAC4), 785 (HDAC5), 1512 (HDAC7), 77 (HDAC8), 2268 (HDAC9), 21 (HDAC10), 22 (HDAC11) | ND | IC50: 0.14 to 0.38 µM (various cancer cell lines) | [74] | |
| Pyrimidinedione derivative (6) | ND | 12.4 | 138 (HDAC1), 444 (HDAC2) | ND | Induces arrest of the cell cycle in subG1 phase and death by apoptosis (colon cancer) | [66][75] | |
| Quinazolin-4-one derivative (3f) | ND | 29 | 65 (HDAC1), 222 (HDAC2), 60 (HDAC18), 141 (HDAC11) | Increases acetylation levels of α-tubulin and histone H3 at 10 μM | ND | [76] | |
| Sulfone derivative (36) | ND | 8 | 138 (HDAC8), 300 (HDAC11) | 0.01 (unspecified) | ND | [66] | |
| Trichostatine A derivatives (M344, 16b) | ND | 88 | 3 (HDAC1) | ND | ND | [77] | |
| Tubacin derivative (WT-161) | Phe200, Phe201, Leu270, Arg194 of HDAC7 | 0.4 | 129 (HDAC3) | 0.3 (multiple myeloma) | IC50 = 3.6 µM (multiple myeloma)SangtingTaoCI50: 1.5 to 4.7 µM (multiple myeloma cell lines) | [78] | |
| Tubastatin A derivative (Marbostat-100) | Asp649, His651 et Asp742 of DD2 | 0.7 | 1106 (HDAC2), 247 (HDAC8) | 0.05 (acute monocytic leukemia) | Non-cytotoxic | [79] | |
| Indolylsulfonylcinnamic hydroxamate (12) | ND | 5.2 | 60 (HDAC1), 223 (HDAC2) | 0.1 (colon cancer) | IC50: 0.4 to 2.5 µM (multiple cancer cell lines) | [80] | |
| MAIP-032 | DD2 | 58 | 38 (HDAC1) | ND | CI50: 3.87 µM (squamous cell carcinoma line of the tongue) | [81] | |
| MPT0G211 | ND | 0.291 | ND | 0.1 (neuroblastoma) | ND | [30] | |
| N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)] (HPB) | His573 and His574 of DD2 | 31 | 37 (HDAC1) | 8 (prostate cancer) | ND | [52][82] | |
| N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide (HPOB) | Binding to zinc ion only via its OH group but does not displace the zinc-bound water molecule | 56 | 52 (HDAC1) | 16 (prostate cancer, adenocarcinoma, glioblastoma) | Increases the effect on cell viability in combination with etoposide, dexamethasone or SAHA | [83][84] | |
| N-hydroxy-4-(2-methoxy-5-(methyl(2-methylquinazolin-4-yl)-amino)phenoxy)butanamide (23bb) | Tyr298 and Glu255 of an HDAC6 homology model | 17 | 25 (HDAC1), 200 (HDAC8) | 0.051 (cervical cancer) | IC50: 14 to 104 nM (various cancer cell lines) | [85] | |
| Nexturastat A | DD2 of an HDAC6 homology model | 5 | 604 (HDAC1) | 0.01 (murine melanoma) | IC50 = 14.3 µM (melanoma) | [57][86] | |
| Oxazole hydroxamate (4g) | Phe620, Phe680, Leu749, and Tyr782 of DD2 of an HDAC6 homology model | 59 | 237 (HDAC1, 8) | 10 (cervical cancer) | IC50 = 10.2 µM (acute myeloid leukemia) | [87] | |
| Ricolinostat (ACY-1215) | DD2 of an HDAC6 homology model | 4.7 | 12 (HDAC1), 10 (HDAC2), 11 (HDAC3), 1490 (HDAC4), 1064 (HDAC5), 298 (HDAC7), 21 (HDAC8), >2000 (HDAC9, 11) | 0.62 (multiple myeloma) | CI50: 2 to 8 µM (multiple myeloma cell lines) | [57][88][89] | |
| Sahaquine | ND | ND | ND | 0.1 (glioblastoma) | CI50: 10 µM (glioblastoma) | [90] | |
| TC24 | Ser568, His610, Phe679 and Tyr782 of HDAC6 | ND | ND | 1 et 10 (gastric cancer) | CI50: 10.2 to 17.2 µM (several gastric cancer cell lines) | [91] | |
| Tetrahydroisoquinoline (5a) | ND | 36 | 1250 (HDAC1), >1000 (HDAC2, 4, 5, 7, 10, 11), 1278 (HDAC3), 58 (HDAC8) | 0.21 (cervical cancer) | ND | [63] | |
| Thiazole | ND | 52 | ND | ND | ND | [63] | |
| Tubacin | DD2 of an HDAC6 homology model | 4 | 350 (HDAC1) | 5 (prostate cancer)SangtingTao2.5 (acute lymphoblastic leukemia) | IC50: 1.2 to 2 µM (acute lymphoblastic leukemia) | [57][92][93] | |
| Tubastatin A | His610, His611, Phe679, Phe680 and Tyr782 of HDAC6 | 15 | 1093 (HDAC1) | 2.5 (unspecified) | ND | [91][92] | |
| Tubathian A | ND | 1.9 | 5790 (HDAC1) | 0.1 (ovarian cancer) | ND | [94] | |
| Other | 3-hydroxypyridine-2-thione (3-HPT) | Tyr306 of HDAC8 | 681 | 5 (HDAC8) | ND | Inactive against two prostate cancer cell lines and one acute T cell leukemia cell line | [95] |
| 1-hydroxypyridine-2-thione (1HPT)-6-carboxylic acid | DD | 150 | 287 (HDAC1), 4733 (HDAC2), 473 (HDAC4), 233 (HDAC5), 1933 (HDAC7), 22 (HDAC8), 313 (HDAC9) | ND | CI50: 18 to 75 µM (leukemia) | [96] | |
| Adamantylamino derivative (20a) | ND | 82 | 46 (HDAC1), 51 (HDAC4) | ND | ND | [97] | |
| Mercaptoacetamide derivative (2b) | ND | 1.3 | 3615 (HDAC1) | 10 (primary rat cortical culture) | ND | [98] | |
| Sulfamide derivative (13e) | ND | 440 | >23 (HDAC1) | 1 (bladder cancer) | ND | [99] | |
| Undefined structure | CKD-506 | ND | 5 | >400 (HDAC1, 2, 7, 8) | 0.03 (Human PBMCs) | ND | [100] |
| HDAC6 Inhibitor | Clinical Trial Identification | Phase of the Clinical Trial | Pathology |
|---|---|---|---|
| ACY-241 | NCT02400242 | Ia/Ib | Multiple myeloma |
| NCT02935790 | Ib | Stage III and IV unresectable melanoma | |
| NCT02551185 | Ib | Advanced solid tumors | |
| NCT02635061 | Ib | Non-resectable non-small cell lung cancer | |
| ACY-1215 | NCT02632071 | Ib | Unresectable or metastatic breast cancer |
| NCT02787369 | Ib | Relapsed chronic lymphocytic leukemia | |
| NCT02091063 | Ib/II | Relapsed or refractory lymphoid malignancies | |
| NCT01997840 | Ib/II | Recurrent and refractory multiple myeloma | |
| NCT01583283 | I/II | Multiple myeloma recurrent or recurrent and refractory | |
| NCT02189343 | Ib | Recurrent and refractory multiple myeloma | |
| NCT01323751 | I/II | Multiple myeloma recurrent or recurrent and refractory | |
| NCT02856568 | Ib | Unresectable or metastatic cholangiocarcinoma | |
| NCT02661815 | Ib | Ovarian cancer, primary peritoneal cancer or platinum-resistant fallopian tubes |
3.2 HDAC6 inhibitors in solid cancers
3.3. HDAC6 inhibitors in hematological malignancies
Similar to pan-HDAC inhibitors approved for the treatment of hematological cancers, specific HDAC6 inhibitors showed anti-cancer properties in various cancer types such as multiple myeloma [110], chronic lymphocytic leukemia [42], acute myeloid leukemia (AML) [111], acute lymphoblastic leukemia (ALL) and chronic myeloid leukemia (CML) [doi: 10.1016/j.phrs.2020.105058].
3.3.1. Nuclear HDAC6 and Its Implication in Leukemia
3.3.2. HDAC6 in CML
3.3.2.1.Degradation of BCR-ABL via Deacetylation of HSP90α by HDAC6 in the Cytoplasm
Although little research exists on HDAC6 in the context of CML, this protein has a function that makes it particularly interesting in the context of such pathology. HDAC6 deacetylates heat shock protein (HSP90)α, which is involved in the stabilization of the oncogenic tyrosine kinase breakpoint cluster region-Abelson (BCR-ABL) protein [113] protein. In the acetylated form, HSP90α loses its chaperone function, which leads to the degradation of its client proteins by the proteasome (Figure 3A). The importance of the acetylation status of HSP90α in the protein degradation of BCR-ABL makes HDAC6 inhibitors potentially promising molecules for the treatment of CML. Pan-HDAC inhibitors are capable of inducing the inhibition of HDAC6, as well as the downregulation of HDAC6 using si-RNA, which increases the acetylation of HSP90α, and in turn increases the ubiquitination of the BCR-ABL protein, decreasing its expression in K562 cells [115][116].
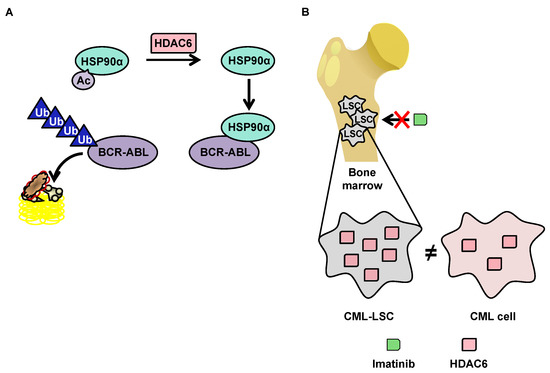
3.3.2.2. OverExpression of HDAC6 in CML Stem Cells
3.3.3 HDAC6 inhibitors in CLL
HDAC6 is upregulated in CLL patient samples, cell lines, and euTCL1 transgenic mouse models compared to normal controls. Accordingly, this pathology could be an interesting target for selective HDAC6 inhibitors, as genetic silencing of HDAC6 improves the survival of euTCL1 mice. Moreover, the chemical inhibitor ACY738 reduces the proliferation of CLL B cells leading to their apoptosis. Together with ibrutinib, this HDAC6 inhibitor triggers synergistic cell death in vivo [119]. Beyond the direct effect on pathological B cells, HDAC6 inhibition improves CLL-induced immunosuppression of CLL T cells. HDAC6 inhibitors enhances immune checkpoint blockade in CLL so that combination treatment with ACY738 potentializes the in vivo antitumor effect of anti-PD-1 and anti-PD-L1 antibody treatments with increased cytotoxic CD8+ T cells [119].
3.3.4 HDAC6 inhibitors in AML
Several HDAC6 inhibitors were assessed as single agents in AML. The HDAC6 inhibitor ST80 shows potent antileukemic activity in myeloid cell lines and primary AML blasts at low micromolar concentrations, leading to preferential acetylation of a-tubulin [120]. HDAC inhibitors with a central naphthoquinone structure selectively inhibit HDAC6 in the AML cell line MV4-11, further decreasing mutant FLT-3 protein and constitutively active signal transducer and activators of transcription (STAT)5 levels and reducing extracellular regulated kinase (ERK) phosphorylation [121]. HDAC inhibitors with the 2-(oxazol-2-yl)phenol moiety as a novel zinc-binding group exhibited selective inhibition against HDAC1 and class IIb HDACs (HDAC6 and HDAC10) in the MV-4-11 AML cells [122]. The in vivo potency of the selective and orally-available HDAC6 inhibitor N-Hydroxy-4-(2-methoxy-5-(methyl(2-methylquinazolin-4-yl)amino)phenoxy)butanamide 23bb was better against MV4-11 AML cells compared to SAHA or ACY-1215 [doi:10.1021/acs.jmedchem.5b01342]. The O-aminobenzamide-based HDAC inhibitor compound 13e down-regulates HDAC6 in MV4-11 cells. 13e induces apoptotic cell death and cycle arrest most likely mediated by a p53-dependent pathway [doi:10.1021/acs.jmedchem.8b00136]. The parthenolide-SAHA hybrid compound 26 more potently reduces the viability of the resistant HL-60/ADR AML cell line compared to SAHA, triggering intrinsic apoptosis and reducing the protein expression levels of HDAC1, HDAC6 and the multidrug resistance-associated protein 1 (ABCC1) leading to an intracellular accumulation of drugs [doi:10.1016/j.bioorg.2019.03.056]. The HDAC6-selective inhibitor PTG-0861 induces apoptosis in MV4-11 AML cells with limited cytotoxicity against non-malignant cells [doi:10.1016/j.ejmech.2020.112411].
In AML, inhibition of HDAC6 was essentially investigated in combination with other pharmacologically active compounds at a pre-clinical level. For instance, a combination of 17-(allylamino)-17-demethoxygeldanamycin (17-AAG), a synthetic derivative of the ansamycin benzoquinone antibiotic geldanamycin, with the HDAC6 inhibitor tubacin reduces the viability of primary AML samples, validating HDAC6 as a HSP90 client protein also in AML and that its hyperacetylation facilitates the anticancer potential of 17-AAG [doi:10.1182/blood-2008-03-143644]. LBH-589 and PXD101 inhibit HDAC1 and HDAC6 and synergize with cytarabine to induce cell death in pediatric AML, accompanied by DNA damage induction and increased Bim expression levels [doi:10.1371/journal.pone.0017138]. Similarly, Bim protein induction and inhibition of nuclear factor-kappa B (NF-kB) pathway were identified as a mechanistic basis for the synergistic anti-cancer effects of belinostat in combination with the proteasome inhibitor bortezomib in AML and ALL cells [doi:10.1111/j.1365-2141.2011.08591.x]. The selective JAK2/HDAC6 dual inhibitor 20a shows excellent in vivo antitumor efficacy in HEL AML mouse xenograft assays and synergizes with the antifungal drug fluconazole [doi:10.1021/acs.jmedchem.8b00393]. The selective HDAC6 inhibitor MPT0G211 combined with doxorubicin displays anti-cancer effect by inducing a DNA damage response associated with increased Ku70 acetylation and BAX activation in HL-60 and MOLT-4 AML cell lines. Accordingly, ectopic expression of HDAC6 successively reverses the apoptosis triggered by the combined treatment [doi:10.1186/s13148-018-0595-8].
3.3.5 HDAC6 inhibitors in ALL
The HDAC6 inhibitor tubacin enhances the anti-cancer effects of the Na+/K+-ATPase inhibitor ouabain or the proteasome inhibitor MG-13 against pre-B and T ALL cells in vitro and in vivo. These results suggest that selectively targeting HDAC6 alone or in combination with conventional chemotherapeutic drugs could provide a novel approach for ALL therapy [doi:10.3109/10428194.2011.570821]. Similarly, belinostat synergizes with the proteasome inhibitor bortezomib to kill ALL cells through Bim up-regulation and NF-kB inhibition. Altogether the perturbation of intracellular microtubular transport network, combined with the interference with protein homeostasis via proteasomal inhibition, could be a general and efficient mechanism explaining the synergistic effect observed [doi:10.1111/j.1365-2141.2011.08591.x]. MPT0G211 combined with vincristine interrupts ALL mitosis via interference with microtubular dynamics leading to apoptosis. In vivo, MPT0G211 plus doxorubicin or vincristine reduces tumor growth xenograft models [doi:10.1186/s13148-018-0595-8].
Remarkably, it has been shown that the inhibition of HDAC6 using either the pan-HDAC inhibitor trichostatin, the selective HDAC6 inhibitor tubacin, or a genetic knock-down efficiently reduces Notch3 signaling through a post-translational-mediated protein down-regulation, leading to enhanced apoptosis in T-ALL cells and impairing leukemia growth in mice xenografted with T-ALL cell lines and primary human T-ALL cells. These results highlights the therapeutic potential of HDAC6 targeting in Notch3-addicted tumors [doi:10.1038/s41388-018-0234-z].
3.3.6 HDAC6 inhibitors in other hematological malignancies
Inhibition of HDAC6 activity increases CD20 levels in B-cell tumor cell lines and malignant patient cells, potentializing the in vivo effect of anti-CD20 monoclonal antibodies like rituximab. Translation of CD20 mRNA is significantly enhanced after HDAC6 inhibition as CD20 mRNA was abundant within the polysomal fraction, indicating a post-transcriptional function of HDAC6. Collectively, these findings suggest HDAC6 inhibition is a rational therapeutic strategy to be implemented in combination therapies with anti-CD20 monoclonal antibodies and open up novel avenues for the clinical use of HDAC6 inhibitors [doi:10.1002/mc.22983].
The HDAC6 inhibitor A452 combined with the Bruton's tyrosine kinase inhibitor ibrutinib efficiently kills non-Hodgkin lymphoma cells, including follicular lymphoma [doi:10.1002/mc.22983].
The HDAC6 inhibitor KT-531 displays the highest anti-cancer potency against T-cell prolymphocytic leukemia (T-PLL) cells compared to other hematological neoplasms, together with safe differential toxicity compared to non-transformed cell lines. Accordingly, HDAC6 is overexpressed in primary T-PLL patient samples in which KT-531 exerts a potent anti-cancer activity. Moreover, a combination of KT-531 with various approved drugs including bendamustine, idasanutlin, and venetoclax shows promising synergistic effects against T-PLL patient cells [doi:10.1021/acs.jmedchem.1c00420].
This entry is adapted from the peer-reviewed paper 10.3390/cancers12020318
References
- Michael Schnekenburger; Cristina Florean; Mario Dicato; Marc Diederich; Epigenetic alterations as a universal feature of cancer hallmarks and a promising target for personalized treatments. Current Topics in Medicinal Chemistry 2015, 16, 745-776, 10.2174/1568026615666150825141330.
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules 2016, 21, 1608.
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193.
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61.
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182.
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100.
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749.
- Bertos, N.R.; Gilquin, B.; Chan, G.K.; Yen, T.J.; Khochbin, S.; Yang, X.J. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J. Biol. Chem. 2004, 279, 48246–48254.
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793.
- Zheng, K.; Jiang, Y.; He, Z.; Kitazato, K.; Wang, Y. Cellular defence or viral assist: The dilemma of HDAC6. J. Gen. Virol. 2017, 98, 322–337.
- Mortenson, J.B.; Heppler, L.N.; Banks, C.J.; Weerasekara, V.K.; Whited, M.D.; Piccolo, S.R.; Johnson, W.E.; Thompson, J.W.; Andersen, J.L. Histone deacetylase 6 (HDAC6) promotes the pro-survival activity of 14-3-3zeta via deacetylation of lysines within the 14-3-3zeta binding pocket. J. Biol. Chem. 2015, 290, 12487–12496.
- Zhang, L.; Liu, S.; Liu, N.; Zhang, Y.; Liu, M.; Li, D.; Seto, E.; Yao, T.P.; Shui, W.; Zhou, J. Proteomic identification and functional characterization of MYH9, Hsc70, and DNAJA1 as novel substrates of HDAC6 deacetylase activity. Protein Cell 2015, 6, 42–54.
- Wu, J.Y.; Xiang, S.; Zhang, M.; Fang, B.; Huang, H.; Kwon, O.K.; Zhao, Y.; Yang, Z.; Bai, W.; Bepler, G.; et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 2018, 293, 1976–1993.
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45.
- Salian-Mehta, S.; Xu, M.; McKinsey, T.A.; Tobet, S.; Wierman, M.E. Novel Interaction of Class IIb Histone Deacetylase 6 (HDAC6) with Class IIa HDAC9 Controls Gonadotropin Releasing Hormone (GnRH) Neuronal Cell Survival and Movement. J. Biol. Chem. 2015, 290, 14045–14056.
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755.
- Medler, T.R.; Craig, J.M.; Fiorillo, A.A.; Feeney, Y.B.; Harrell, J.C.; Clevenger, C.V. HDAC6 Deacetylates HMGN2 to Regulate Stat5a Activity and Breast Cancer Growth. Mol. Cancer Res. 2016, 14, 994–1008.
- Chang, Y.W.; Tseng, C.F.; Wang, M.Y.; Chang, W.C.; Lee, C.C.; Chen, L.T.; Hung, M.C.; Su, J.L. Deacetylation of HSPA5 by HDAC6 leads to GP78-mediated HSPA5 ubiquitination at K447 and suppresses metastasis of breast cancer. Oncogene 2016, 35, 1517–1528.
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072–1077.
- Liu, K.P.; Zhou, D.; Ouyang, D.Y.; Xu, L.H.; Wang, Y.; Wang, L.X.; Pan, H.; He, X.H. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 2013, 441, 970–975.
- Zhang, M.; Xiang, S.; Joo, H.Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C.; et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSalpha. Mol. Cell 2014, 55, 31–46.
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445.
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638.
- Moreno-Gonzalo, O.; Mayor, F., Jr.; Sanchez-Madrid, F. HDAC6 at Crossroads of Infection and Innate Immunity. Trends Immunol. 2018, 39, 591–595.
- Nakka, K.K.; Chaudhary, N.; Joshi, S.; Bhat, J.; Singh, K.; Chatterjee, S.; Malhotra, R.; De, A.; Santra, M.K.; Dilworth, F.J.; et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc. Natl. Acad. Sci. USA 2015, 112, E3374–E3383.
- Huo, L.; Li, D.; Sun, X.; Shi, X.; Karna, P.; Yang, W.; Liu, M.; Qiao, W.; Aneja, R.; Zhou, J. Regulation of Tat acetylation and transactivation activity by the microtubule-associated deacetylase HDAC6. J. Biol. Chem. 2011, 286, 9280–9286.
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831.
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118.
- Haakenson, J.; Zhang, X. HDAC6 and ovarian cancer. Int. J. Mol. Sci. 2013, 14, 9514–9535.
- Fan, S.J.; Huang, F.I.; Liou, J.P.; Yang, C.R. The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis. 2018, 9, 655.
- Lee, S.W.; Yang, J.; Kim, S.Y.; Jeong, H.K.; Lee, J.; Kim, W.J.; Lee, E.J.; Kim, H.S. MicroRNA-26a induced by hypoxia targets HDAC6 in myogenic differentiation of embryonic stem cells. Nucleic Acids Res. 2015, 43, 2057–2073.
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, F., Jr.; Penela, P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2012, 31, 856–869.
- Williams, K.A.; Zhang, M.; Xiang, S.; Hu, C.; Wu, J.Y.; Zhang, S.; Ryan, M.; Cox, A.D.; Der, C.J.; Fang, B.; et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 2013, 288, 33156–33170.
- Di Fulvio, S.; Azakir, B.A.; Therrien, C.; Sinnreich, M. Dysferlin interacts with histone deacetylase 6 and increases alpha-tubulin acetylation. PLoS ONE 2011, 6, e28563.
- Tala, S.X.; Chen, J.; Zhang, L.; Liu, N.; Zhou, J.; Li, D.; Liu, M. Microtubule stabilization by Mdp3 is partially attributed to its modulation of HDAC6 in addition to its association with tubulin and microtubules. PLoS ONE 2014, 9, e90932.
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016.
- Salemi, L.M.; Almawi, A.W.; Lefebvre, K.J.; Schild-Poulter, C. Aggresome formation is regulated by RanBPM through an interaction with HDAC6. Biol. Open 2014, 3, 418–430.
- Perez, M.; Santa-Maria, I.; Gomez de Barreda, E.; Zhu, X.; Cuadros, R.; Cabrero, J.R.; Sanchez-Madrid, F.; Dawson, H.N.; Vitek, M.P.; Perry, G.; et al. Tau--an inhibitor of deacetylase HDAC6 function. J. Neurochem. 2009, 109, 1756–1766.
- Schofield, A.V.; Gamell, C.; Bernard, O. Tubulin polymerization promoting protein 1 (TPPP1) increases beta-catenin expression through inhibition of HDAC6 activity in U2OS osteosarcoma cells. Biochem. Biophys. Res. Commun. 2013, 436, 571–577.
- Batchu, S.N.; Brijmohan, A.S.; Advani, A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin. Sci. (Lond.) 2016, 130, 987–1003.
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111.
- Maharaj, K.; Powers, J.J.; Achille, A.; Deng, S.; Fonseca, R.; Pabon-Saldana, M.; Quayle, S.N.; Jones, S.S.; Villagra, A.; Sotomayor, E.M.; et al. Silencing of HDAC6 as a therapeutic target in chronic lymphocytic leukemia. Blood Adv. 2018, 2, 3012–3024.
- Li, A.; Chen, P.; Leng, Y.; Kang, J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene 2018, 37, 5952–5966.
- Qian, H.; Chen, Y.; Nian, Z.; Su, L.; Yu, H.; Chen, F.J.; Zhang, X.; Xu, W.; Zhou, L.; Liu, J.; et al. HDAC6-mediated acetylation of lipid droplet-binding protein CIDEC regulates fat-induced lipid storage. J. Clin. Invest. 2017, 127, 1353–1369.
- Bamodu, O.A.; Kuo, K.T.; Yuan, L.P.; Cheng, W.H.; Lee, W.H.; Ho, Y.S.; Chao, T.Y.; Yeh, C.T. HDAC inhibitor suppresses proliferation and tumorigenicity of drug-resistant chronic myeloid leukemia stem cells through regulation of hsa-miR-196a targeting BCR/ABL1. Exp. Cell Res. 2018, 370, 519–530.
- Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337.
- Dehmel, F.; Weinbrenner, S.; Julius, H.; Ciossek, T.; Maier, T.; Stengel, T.; Fettis, K.; Burkhardt, C.; Wieland, H.; Beckers, T. Trithiocarbonates as a novel class of HDAC inhibitors: SAR studies, isoenzyme selectivity, and pharmacological profiles. J. Med. Chem. 2008, 51, 3985–4001.
- Inks, E.S.; Josey, B.J.; Jesinkey, S.R.; Chou, C.J. A novel class of small molecule inhibitors of HDAC6. ACS Chem. Biol. 2012, 7, 331–339.
- Schafer, S.; Saunders, L.; Eliseeva, E.; Velena, A.; Jung, M.; Schwienhorst, A.; Strasser, A.; Dickmanns, A.; Ficner, R.; Schlimme, S.; et al. Phenylalanine-containing hydroxamic acids as selective inhibitors of class IIb histone deacetylases (HDACs). Bioorg. Med. Chem. 2008, 16, 2011–2033.
- Schafer, S.; Saunders, L.; Schlimme, S.; Valkov, V.; Wagner, J.M.; Kratz, F.; Sippl, W.; Verdin, E.; Jung, M. Pyridylalanine-containing hydroxamic acids as selective HDAC6 inhibitors. ChemMedChem 2009, 4, 283–290.
- Jochems, J.; Boulden, J.; Lee, B.G.; Blendy, J.A.; Jarpe, M.; Mazitschek, R.; Van Duzer, J.H.; Jones, S.; Berton, O. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 2014, 39, 389–400.
- Porter, N.J.; Mahendran, A.; Breslow, R.; Christianson, D.W. Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464.
- Krukowski, K.; Ma, J.; Golonzhka, O.; Laumet, G.O.; Gutti, T.; van Duzer, J.H.; Mazitschek, R.; Jarpe, M.B.; Heijnen, C.J.; Kavelaars, A. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 2017, 158, 1126–1137.
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [(18)F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014.
- Wagner, F.F.; Olson, D.E.; Gale, J.P.; Kaya, T.; Weiwer, M.; Aidoud, N.; Thomas, M.; Davoine, E.L.; Lemercier, B.C.; Zhang, Y.L.; et al. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem. 2013, 56, 1772–1776.
- Kozikowski, A.P.; Tapadar, S.; Luchini, D.N.; Kim, K.H.; Billadeau, D.D. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: A new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J. Med. Chem. 2008, 51, 4370–4373.
- Sixto-Lopez, Y.; Bello, M.; Rodriguez-Fonseca, R.A.; Rosales-Hernandez, M.C.; Martinez-Archundia, M.; Gomez-Vidal, J.A.; Correa-Basurto, J. Searching the conformational complexity and binding properties of HDAC6 through docking and molecular dynamic simulations. J. Biomol. Struct. Dyn. 2017, 35, 2794–2814.
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707.
- Olsen, C.A.; Ghadiri, M.R. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic alpha3beta-tetrapeptides. J. Med. Chem. 2009, 52, 7836–7846.
- Chen, Y.; Lopez-Sanchez, M.; Savoy, D.N.; Billadeau, D.D.; Dow, G.S.; Kozikowski, A.P. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J. Med. Chem. 2008, 51, 3437–3448.
- Diedrich, D.; Hamacher, A.; Gertzen, C.G.; Alves Avelar, L.A.; Reiss, G.J.; Kurz, T.; Gohlke, H.; Kassack, M.U.; Hansen, F.K. Rational design and diversity-oriented synthesis of peptoid-based selective HDAC6 inhibitors. Chem. Commun. (Camb.) 2016, 52, 3219–3222.
- Lin, X.; Chen, W.; Qiu, Z.; Guo, L.; Zhu, W.; Li, W.; Wang, Z.; Zhang, W.; Zhang, Z.; Rong, Y.; et al. Design and synthesis of orally bioavailable aminopyrrolidinone histone deacetylase 6 inhibitors. J. Med. Chem. 2015, 58, 2809–2820.
- Blackburn, C.; Barrett, C.; Chin, J.; Garcia, K.; Gigstad, K.; Gould, A.; Gutierrez, J.; Harrison, S.; Hoar, K.; Lynch, C.; et al. Potent histone deacetylase inhibitors derived from 4-(aminomethyl)-N-hydroxybenzamide with high selectivity for the HDAC6 isoform. J. Med. Chem. 2013, 56, 7201–7211.
- Seidel, C.; Schnekenburger, M.; Mazumder, A.; Teiten, M.H.; Kirsch, G.; Dicato, M.; Diederich, M. 4-Hydroxybenzoic acid derivatives as HDAC6-specific inhibitors modulating microtubular structure and HSP90alpha chaperone activity against prostate cancer. Biochem. Pharmacol. 2016, 99, 31–52.
- Tang, G.; Wong, J.C.; Zhang, W.; Wang, Z.; Zhang, N.; Peng, Z.; Zhang, Z.; Rong, Y.; Li, S.; Zhang, M.; et al. Identification of a novel aminotetralin class of HDAC6 and HDAC8 selective inhibitors. J. Med. Chem. 2014, 57, 8026–8034.
- Wang, X.X.; Wan, R.Z.; Liu, Z.P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418.
- Liang, T.; Hou, X.; Zhou, Y.; Yang, X.; Fang, H. Design, Synthesis, and Biological Evaluation of 2,4-Imidazolinedione Derivatives as HDAC6 Isoform-Selective Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1122–1127.
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.; Chen, B.; D’Annibale, M.A.; Suto, C.M.; Langley, B.C. Functional differences in epigenetic modulators-superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J. Med. Chem. 2007, 50, 3054–3061.
- Lee, H.Y.; Nepali, K.; Huang, F.I.; Chang, C.Y.; Lai, M.J.; Li, Y.H.; Huang, H.L.; Yang, C.R.; Liou, J.P. (N-Hydroxycarbonylbenylamino)quinolines as Selective Histone Deacetylase 6 Inhibitors Suppress Growth of Multiple Myeloma in Vitro and in Vivo. J. Med. Chem. 2018, 61, 905–917.
- Shen, S.; Hadley, M.; Ustinova, K.; Pavlicek, J.; Knox, T.; Noonepalle, S.; Tavares, M.T.; Zimprich, C.A.; Zhang, G.; Robers, M.B.; et al. Discovery of a New Isoxazole-3-hydroxamate-Based Histone Deacetylase 6 Inhibitor SS-208 with Antitumor Activity in Syngeneic Melanoma Mouse Models. J. Med. Chem. 2019, 62, 8557–8577.
- Vogerl, K.; Ong, N.; Senger, J.; Herp, D.; Schmidtkunz, K.; Marek, M.; Muller, M.; Bartel, K.; Shaik, T.B.; Porter, N.J.; et al. Synthesis and Biological Investigation of Phenothiazine-Based Benzhydroxamic Acids as Selective Histone Deacetylase 6 Inhibitors. J. Med. Chem. 2019, 62, 1138–1166.
- Porter, N.J.; Osko, J.D.; Diedrich, D.; Kurz, T.; Hooker, J.M.; Hansen, F.K.; Christianson, D.W. Histone Deacetylase 6-Selective Inhibitors and the Influence of Capping Groups on Hydroxamate-Zinc Denticity. J. Med. Chem. 2018, 61, 8054–8060.
- Duan, W.; Li, J.; Inks, E.S.; Chou, C.J.; Jia, Y.; Chu, X.; Li, X.; Xu, W.; Zhang, Y. Design, synthesis, and antitumor evaluation of novel histone deacetylase inhibitors equipped with a phenylsulfonylfuroxan module as a nitric oxide donor. J. Med. Chem. 2015, 58, 4325–4338.
- Cho, M.; Choi, E.; Yang, J.S.; Lee, C.; Seo, J.J.; Kim, B.S.; Oh, S.J.; Kim, H.M.; Lee, K.; Park, S.K.; et al. Discovery of pyridone-based histone deacetylase inhibitors: Approaches for metabolic stability. ChemMedChem 2013, 8, 272–279.
- Liu, Y.M.; Lee, H.Y.; Lai, M.J.; Pan, S.L.; Huang, H.L.; Kuo, F.C.; Chen, M.C.; Liou, J.P. Pyrimidinedione-mediated selective histone deacetylase 6 inhibitors with antitumor activity in colorectal cancer HCT116 cells. Org. Biomol. Chem. 2015, 13, 10226–10235.
- Yu, C.W.; Chang, P.T.; Hsin, L.W.; Chern, J.W. Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 6775–6791.
- Heltweg, B.; Dequiedt, F.; Marshall, B.L.; Brauch, C.; Yoshida, M.; Nishino, N.; Verdin, E.; Jung, M. Subtype selective substrates for histone deacetylases. J. Med. Chem. 2004, 47, 5235–5243.
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167.
- Sellmer, A.; Stangl, H.; Beyer, M.; Grunstein, E.; Leonhardt, M.; Pongratz, H.; Eichhorn, E.; Elz, S.; Striegl, B.; Jenei-Lanzl, Z.; et al. Marbostat-100 Defines a New Class of Potent and Selective Antiinflammatory and Antirheumatic Histone Deacetylase 6 Inhibitors. J. Med. Chem. 2018, 61, 3454–3477.
- Lee, H.Y.; Tsai, A.C.; Chen, M.C.; Shen, P.J.; Cheng, Y.C.; Kuo, C.C.; Pan, S.L.; Liu, Y.M.; Liu, J.F.; Yeh, T.K.; et al. Azaindolylsulfonamides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer HCT116 cells. J. Med. Chem. 2014, 57, 4009–4022.
- Mackwitz, M.K.W.; Hamacher, A.; Osko, J.D.; Held, J.; Scholer, A.; Christianson, D.W.; Kassack, M.U.; Hansen, F.K. Multicomponent Synthesis and Binding Mode of Imidazo[1,2 -a]pyridine-Capped Selective HDAC6 Inhibitors. Org. Lett. 2018, 20, 3255–3258.
- Lee, J.H.; Yao, Y.; Mahendran, A.; Ngo, L.; Venta-Perez, G.; Choy, M.L.; Breslow, R.; Marks, P.A. Creation of a histone deacetylase 6 inhibitor and its biological effects [corrected]. Proc. Natl. Acad. Sci. USA 2015, 112, 12005–12010.
- Lee, J.H.; Mahendran, A.; Yao, Y.; Ngo, L.; Venta-Perez, G.; Choy, M.L.; Kim, N.; Ham, W.S.; Breslow, R.; Marks, P.A. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. USA 2013, 110, 15704–15709.
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747.
- Yang, Z.; Wang, T.; Wang, F.; Niu, T.; Liu, Z.; Chen, X.; Long, C.; Tang, M.; Cao, D.; Wang, X.; et al. Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem. 2016, 59, 1455–1470.
- Bergman, J.A.; Woan, K.; Perez-Villarroel, P.; Villagra, A.; Sotomayor, E.M.; Kozikowski, A.P. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem. 2012, 55, 9891–9899.
- Senger, J.; Melesina, J.; Marek, M.; Romier, C.; Oehme, I.; Witt, O.; Sippl, W.; Jung, M. Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors. J. Med. Chem. 2016, 59, 1545–1555.
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589.
- Wang, F.; Zhong, B.W.; Zhao, Z.R. ACY 1215, a histone deacetylase 6 inhibitor, inhibits cancer cell growth in melanoma. J. Biol. Regul. Homeost. Agents 2018, 32, 851–858.
- Zhang, I.; Beus, M.; Stochaj, U.; Le, P.U.; Zorc, B.; Rajic, Z.; Petrecca, K.; Maysinger, D. Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule. Cell Death Discov 2018, 4, 41.
- Dong, J.; Zheng, N.; Wang, X.; Tang, C.; Yan, P.; Zhou, H.B.; Huang, J. A novel HDAC6 inhibitor exerts an anti-cancer effect by triggering cell cycle arrest and apoptosis in gastric cancer. Eur. J. Pharmacol. 2018, 828, 67–79.
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846.
- Aldana-Masangkay, G.I.; Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Hsieh, Y.T.; Kim, Y.M.; Lomenick, B.; Okemoto, K.; Landaw, E.M.; Wang, D.; et al. Tubacin suppresses proliferation and induces apoptosis of acute lymphoblastic leukemia cells. Leuk. Lymphoma 2011, 52, 1544–1555.
- Depetter, Y.; Geurs, S.; De Vreese, R.; Goethals, S.; Vandoorn, E.; Laevens, A.; Steenbrugge, J.; Meyer, E.; de Tullio, P.; Bracke, M.; et al. Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer 2019, 145, 735–747.
- Patil, V.; Sodji, Q.H.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition. J. Med. Chem. 2013, 56, 3492–3506.
- Muthyala, R.; Shin, W.S.; Xie, J.; Sham, Y.Y. Discovery of 1-hydroxypyridine-2-thiones as selective histone deacetylase inhibitors and their potential application for treating leukemia. Bioorg. Med. Chem. Lett. 2015, 25, 4320–4324.
- Itoh, Y.; Suzuki, T.; Kouketsu, A.; Suzuki, N.; Maeda, S.; Yoshida, M.; Nakagawa, H.; Miyata, N. Design, synthesis, structure--selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J. Med. Chem. 2007, 50, 5425–5438.
- Segretti, M.C.; Vallerini, G.P.; Brochier, C.; Langley, B.; Wang, L.; Hancock, W.W.; Kozikowski, A.P. Thiol-Based Potent and Selective HDAC6 Inhibitors Promote Tubulin Acetylation and T-Regulatory Cell Suppressive Function. ACS Med. Chem. Lett. 2015, 6, 1156–1161.
- Wahhab, A.; Smil, D.; Ajamian, A.; Allan, M.; Chantigny, Y.; Therrien, E.; Nguyen, N.; Manku, S.; Leit, S.; Rahil, J.; et al. Sulfamides as novel histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 336–340.
- Choi, E.W.; Song, J.W.; Ha, N.; Choi, Y.I.; Kim, S. CKD-506, a novel HDAC6-selective inhibitor, improves renal outcomes and survival in a mouse model of systemic lupus erythematosus. Sci. Rep. 2018, 8, 17297.
- Schnekenburger, M.; Florean, C.; Dicato, M.; Diederich, M. Epigenetic alterations as a universal feature of cancer hallmarks and a promising target for personalized treatments. Curr. Top. Med. Chem. 2016, 16, 745–776.
- Rosik, L.; Niegisch, G.; Fischer, U.; Jung, M.; Schulz, W.A.; Hoffmann, M.J. Limited efficacy of specific HDAC6 inhibition in urothelial cancer cells. Cancer Biol. Ther. 2014, 15, 742–757.
- Hsieh, Y.L.; Tu, H.J.; Pan, S.L.; Liou, J.P.; Yang, C.R. Anti-metastatic activity of MPT0G211, a novel HDAC6 inhibitor, in human breast cancer cells in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 992–1003.
- Shan, B.; Yao, T.P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283, 21065–21073.
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rossig, L.; Seto, E.; Augustin, H.G.; et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011, 30, 4142–4156.
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107.
- Valdespino, V.; Valdespino, P.M. Potential of epigenetic therapies in the management of solid tumors. Cancer Manag. Res. 2015, 7, 241–251.
- Grassadonia, A.; Cioffi, P.; Simiele, F.; Iezzi, L.; Zilli, M.; Natoli, C. Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies. Cancers 2013, 5, 919–942.
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.M.; Tian, X.; Wang, A.Z. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215.
- Brindisi, M.; Saraswati, A.P.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. J. Med. Chem. 2020, 63, 23–39.
- Hackanson, B.; Rimmele, L.; Benkisser, M.; Abdelkarim, M.; Fliegauf, M.; Jung, M.; Lubbert, M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk. Res. 2012, 36, 1055–1062.
- Liu, Y.; Peng, L.; Seto, E.; Huang, S.; Qiu, Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 2012, 287, 29168–29174.
- Kramer, O.H.; Mahboobi, S.; Sellmer, A. Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 2014, 35, 501–509.
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171.
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734.
- Rao, R.; Fiskus, W.; Yang, Y.; Lee, P.; Joshi, R.; Fernandez, P.; Mandawat, A.; Atadja, P.; Bradner, J.E.; Bhalla, K. HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008, 112, 1886–1893.
- Chen, P.B.; Hung, J.H.; Hickman, T.L.; Coles, A.H.; Carey, J.F.; Weng, Z.; Chu, F.; Fazzio, T.G. Hdac6 regulates Tip60-p400 function in stem cells. Elife 2013, 2, e01557.
- Kuo, Y.H.; Qi, J.; Cook, G.J. Regain control of p53: Targeting leukemia stem cells by isoform-specific HDAC inhibition. Exp. Hematol. 2016, 44, 315–321.
- Kamira Maharaj; John J. Powers; Melanie Mediavilla-Varela; Alex Achille; Wael Gamal; Steven Quayle; Simon S. Jones; Eva Sahakian; Javier Pinilla-Ibarz; HDAC6 Inhibition Alleviates CLL-Induced T-Cell Dysfunction and Enhances Immune Checkpoint Blockade Efficacy in the Eμ-TCL1 Model. Frontiers in Immunology 2020, 11, -, 10.3389/fimmu.2020.590072.
- Björn Hackanson; Leander Rimmele; Marco Benkißer; Mahmoud Abdelkarim; Manfred Fliegauf; Manfred Jung; Michael Lübbert; HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leukemia Research 2012, 36, 1055-1062, 10.1016/j.leukres.2012.02.026.
- Elizabeth S. Inks; Benjamin J. Josey; Sean R. Jesinkey; C. James Chou; A Novel Class of Small Molecule Inhibitors of HDAC6. ACS Chemical Biology 2011, 7, 331-339, 10.1021/cb200134p.
- Youxuan Li; Patrick M. Woster; Discovery of a new class of histone deacetylase inhibitors with a novel zinc binding group. MedChemComm 2014, 6, 613-618, 10.1039/c4md00401a.