Understanding the autistic brain and the involvement of genetic, non-genetic, and numerous signaling pathways in the etiology and pathophysiology of autism spectrum disorder (ASD) is complex, as is evident from various studies. Apart from multiple developmental disorders of the brain, autistic subjects show a few characteristics like impairment in social communications related to repetitive, restricted, or stereotypical behavior, which suggests alterations in neuronal circuits caused by defects in various signaling pathways during embryogenesis. Most of the research studies on ASD subjects and genetic models revealed the involvement of mutated genes with alterations of numerous signaling pathways like Wnt, hedgehog, and Retinoic Acid (RA).
- autism spectrum disorder
- Asperger’s syndrome
- neuropathological alterations
- hedgehog signaling pathway
1. Introduction
2. Types of Autism Spectrum Disorder
|
Levels |
Clinical Symptoms |
||
|---|---|---|---|
|
Social Communication |
Repetitive Behavior |
||
|
Level 1 |
Requires extensive medical support |
Severe impairment in verbal and non-verbal communication; a deficit in social interactions; less response to social overtures (e.g., rarely starts an interaction if they have some words of intelligible speech and respond only to direct social overtures. |
Rigid behavior; extreme problems coping with change; repetitive or restricted behavior marked by interferences in body functioning in all spheres; great difficulty changing action or focus. |
|
Level 2 |
Requires medical support |
Marked impairment in verbal and non-verbal communication; a deficit in social interaction even with support; minimum responses to social overtures like simple spoken sentences; less interest in interaction, and odd behavior in non-verbal communication |
Rigid behavior; difficulty coping with change, repetitive or restricted behavior affecting various functions in different contexts; trouble changing action or focus. |
|
Level 3 |
Requires support |
Noticeable impairments in social interaction without support; problems initiating interactions with people and appears to have less interest in social communication (e.g., affected person speaks full sentences with others but to-and-fro conversation fails and attempts to make friends typically not successful and odd), |
Rigid behavior causes difficulty with functioning in several contexts; problems switching from one activity to another; a deficit in behavior while organizing and planning inhibits independence. |
3. Etiology and Pathophysiology of ASD
4. Impairment of Developmental Pathways
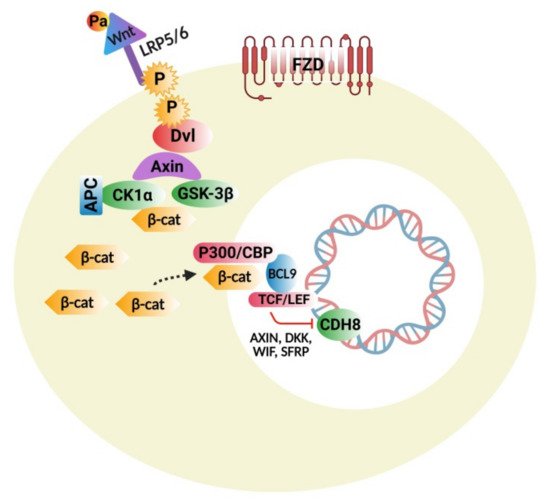
4.1. Wnt Protein and β-Catenin Signaling Pathways
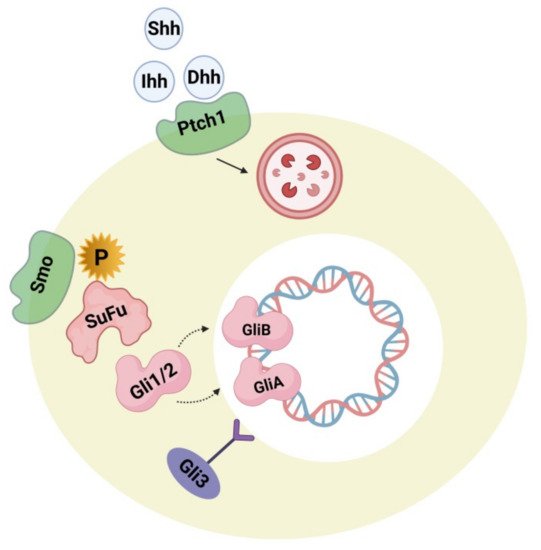
4.2. Hedgehog Signaling Pathway
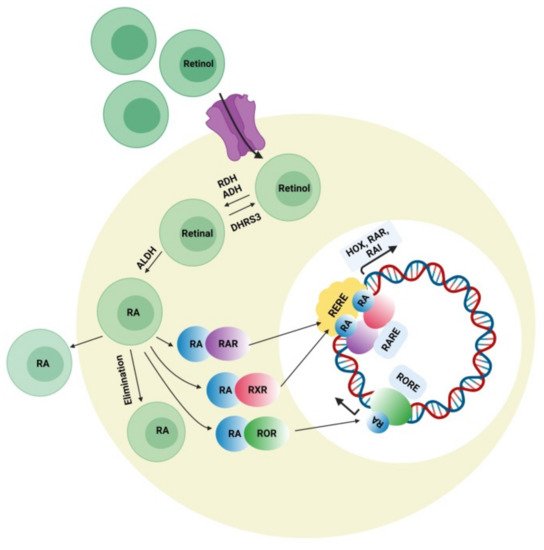
4.3. Retinoic Acid (RA) Signaling Pathway
Retinoic acid can affect various developmental genes that contain retinoic acid response elements (RARE) together with their regulatory spaces. This role of RA suggests interconnections among neurodevelopmental disorders and RA signaling pathways. During embryonic development, retinoic acid helps regulate a set of HOX (homeobox) genes that, during embryogenesis, shapes the upper-body pattern both anteriorly and posteriorly and is involved in brain patterning. RA is engaged in neural-cell differentiation involving dopaminergic and GABAergic neurons. It is also a key induction component (together with sonic hedgehog) for the differentiation of motor neurons from pluripotent stem cells [44]. Moreover retinoic acid is also important for the normal functioning of motor neurons. It was also reported that RA is important for neural migration and neurogenesis in the granular zone of the hippocampus, sub-ventricular zone, and olfactory bulb [45]. Retinol concentration in adequate amount is required for normal functioning of RA signaling pathway presuming that all the enzymes related to RA pathway and nuclear factors work accordingly. Figure 3 represents the synthesis of retinoic acid and the retinoic acid signaling pathway. The deficiency of retinol is one of the major causes of decreased intracellular RA signaling. Some studies demonstrated that the deficiency of retinol in rats during pregnancy decreases RA receptor expression (RAR, beta isoform) in the hypothalamus, causing autistic-like symptoms in the neonates [46]. Interestingly, a lower level of retinol was detected in some autistic subjects when compared with the normal control group in China, which was possibly a synergistic factor in ASD symptom development [47]. Retinol supplements can activate RAR expression and lessen ASD symptoms. The important metabolic step in the RA pathway is the conversion of retinal to retinoic acid with the help of the enzyme (ALDH) retinaldehyde dehydrogenase, which ensures the concentration of RA in the cell. An increase in the degradation rate of this enzyme’s isoform ALDH 1A2 because of over-ubiquitinoylation by the enzyme UBE3A (ubiquitin ligase E3) was established in vitro, and autistic features were observed in mice with an overexpression of UBE3A [48]. The loss of function of UBE3A is related to a neurodevelopmental disorder (Angelman syndrome) showing its importance in brain development [49]. The nuclear receptors for RA have also been involved in ASD pathology. RORs (retinoic acid related orphan receptors) activate transcription of several genes by acting as transcriptional regulators upon retinoic acid-binding. A reduction in ROR gene expression due to hypermethylation was found in autistic subjects. In addition, an immunohistochemical analysis of the postmortem brains of autistic subjects confirmed a lower level of ROR alpha protein [50]. Disruption of the retinoic acid enzymatic production pathway was found to be associated with ASD phenotypes and retinoic acid nuclear receptors, which have also been involved in the pathophysiology of ASD; hence, additional studies have to be performed to establish the correlation between ASD pathogenesis and the involvement of RAR and ROR agonists for autism treatment. Quantitative EEG analysis is another signal-detection tool for diagnosing ASD. The details of the individual characterization of EEG fluctuations in ASD subjects could help examine issues of the brain, which would be useful for observing automatic groupings and random draws of the patient population when analyzing the sensory-processing issues of the brain and the peripheral system [51].
This entry is adapted from the peer-reviewed paper 10.3390/cells10040958
References
- Zhang, X.; Lv, C.-C.; Tian, J.; Miao, R.-J.; Xi, W.; Hertz-Picciotto, I.; Qi, L. Prenatal and Perinatal Risk Factors for Autism in China. J. Autism Dev. Disord. 2010, 40, 1311–1321.
- Baxter, A.J.; Brugha, T.S.; Erskine, H.E.; Scheurer, R.W.; Vos, T.; Scott, J.G. The epidemiology and global burden of autism spectrum disorders. Psychol. Med. 2015, 45, 601–613.
- Daley, T.C. From symptom recognition to diagnosis: Children with autism in urban India. Soc. Sci. Med. 2004, 58, 1323–1335.
- Fusar-Poli, L.; Brondino, N.; Politi, P.; Aguglia, E. Missed diagnoses and misdiagnoses of adults with autism spectrum disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 1–12.
- Vismara, L.A.; Rogers, S.J. Behavioral Treatments in Autism Spectrum Disorder: What Do We Know? Annu. Rev. Clin. Psychol. 2010, 6, 447–468.
- Johnson, C.P.; Myers, S.M.; American Academy of Pediatrics, the Council on Children with Disabilities. Identification and Evaluation of Children with Autism Spectrum Disorders. Pediatrics 2007, 120, 1183–1215.
- Chakrabarti, S.; Fombonne, E. Pervasive developmental disorders in preschool children. JAMA J. Am. Med Assoc. 2001, 285, 3093–3099.
- Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorders—Autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12.
- Karande, S. Autism: A review for family physicians. Indian J. Med Sci. 2006, 60, 205–215.
- Krishnamurthy, V. A Clinical Experience of Autism in India. J. Dev. Behav. Pediatr. 2008, 29, 331–333.
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474.
- Zeldovich, L. The Evolution of ‘Autism’ as a Diagnosis, Explained. 2018. Available online: (accessed on 31 January 2021).
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; (DSM-5); American Psychiatric Association: Washington, DC, USA, 2013; p. 5.
- Asperger’s Syndrome. Available online: (accessed on 28 January 2021).
- Centre for Disease Control and Prevention (CDC). Autism Spectrum Disorder. Available online: (accessed on 13 April 2021).
- Laurvick, C.L.; de Klerk, N.; Bower, C.; Christodoulou, J.; Ravine, D.; Ellaway, C.; Williamson, S.; Leonard, H. Rett syndrome in Australia: A review of the epidemiology. J. Pediatr. 2006, 148, 347–352.
- Volkmar, F.R.; Reichow, B.; McPartland, J. Classification of autism and related conditions: Progress, challenges, and opportunities. Dialog. Clin. Neurosci. 2012, 14, 229–237.
- Walker, D.R.; Thompson, A.; Zwaigenbaum, L.; Goldberg, J.; Bryson, S.E.; Mahoney, W.J.; Strawbridge, C.P.; Szatmari, P. Specifying PDD-NOS: A Comparison of PDD-NOS, Asperger Syndrome, and Autism. J. Am. Acad. Child Adolesc. Psychiatry 2004, 43, 172–180.
- Karabekiroglu, K. Pervasive Developmental Disorder- not Otherwise Specified: Specifying and Differentiating. In Autism Spectrum Disorders: The Role of Genetics in Diagnosis and Treatment; IntechOpen: London, UK, 2011.
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174.
- Zwaigenbaum, L.; Szatmari, P.; Bryson, S.E.; MacLean, J.E.; Tuff, L.P.; Bartolucci, G.; Tuff, L. Pregnancy and birth com-plications in autism and liability to the broader autism phenotype. J. Am. Acad. Child. Adolesc. Psychiatry 2002, 41, 572–579.
- Folstein, S.; Rutter, M. Infantile autism: A genetic study of 21 twin pairs. J. Child Psychol. Psychiatry 1977, 18, 297–321.
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77.
- Casanova, M.F.; Buxhoeveden, D.P.; Switala, A.E.; Roy, E. Minicolumnar pathology in autism. Neurology 2002, 58, 428–432.
- Critchley, H.D.; Daly, E.M.; Bullmore, E.T.; Williams, S.C.; Van Amelesvoort, T.; Robertson, D.M.; Rowe, A.; Phillips, M.; McAlonan, G.; Howlin, P.; et al. The functional neuroanatomy of social behaviour: Changes in cerebral blood flow in people with autistic disorder process facial expressions. Brain 2000, 123, 2203–2212.
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012.
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398.
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75.
- Fuccillo, M.V.; Joyner, A.L.; Fishell, G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 772–783.
- Álvarez-Buylla, A.; Ihrie, R.A. Sonic hedgehog signaling in the postnatal brain. Semin. Cell Dev. Biol. 2014, 33, 105–111.
- Hill, S.A.; Blaeser, A.S.; Coley, A.A.; Xie, Y.; Shepard, K.A.; Harwell, C.C.; Gao, W.-J.; Garcia, A.D.R. Sonic hedgehog signaling in astrocytes mediates cell type-specific synaptic organization. eLife 2019, 8.
- Kumar, S.; Reynolds, K.; Ji, Y. Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 2019, 11, 1–14.
- Al-Ayadhi, L.Y. Relationship between Sonic Hedgehog Protein, Brain-Derived Neurotrophic Factor and Oxidative Stress in Autism Spectrum Disorders. Neurochem. Res. 2011, 37, 394–400.
- Ghanizadeh, A. Malondialdehyde, Bcl-2, Superoxide Dismutase and Glutathione Peroxidase may Mediate the Association of Sonic Hedgehog Protein and Oxidative Stress in Autism. Neurochem. Res. 2012, 37, 899–901.
- Bashir, S.; Halepoto, D.M.; Al-Ayadhi, L. Serum level of desert hedgehog protein in autism spectrum disorder: Preliminary results. Med. Princ. Pr. 2014, 23, 14–17.
- Halepoto, D.M.; Bashir, S.; Zeina, R.; Al-Ayadhi, L.Y. Correlation between Hedgehog (Hh) Protein Family and Brain-Derived Neurotrophic Factor (BDNF) in Autism Spectrum Disorder (ASD). J. Coll. Physicians Surg. Pak. 2015, 25, 882–885.
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510.
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The Crosstalk of mTOR/S6K1 and Hedgehog Pathways. Cancer Cell 2012, 21, 374–387.
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2016, 8, 833–845.
- Goldani, A.A.S.; Downs, S.R.; Widjaja, F.; Lawton, B.; Hendren, R.L. Biomarkers in Autism. Front. Psychiatry 2014, 5, 100.
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-Derived Hedgehog Proteins Modulate Th Differentiation and Disease. J. Immunol. 2013, 190, 2641–2649.
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939.
- Ohja, K.; Gozal, E.; Fahnestock, M.; Cai, L.; Cai, J.; Freedman, J.H.; Switala, A.; El-Baz, A.; Barnes, G.N. Neuroimmunologic and Neurotrophic Interactions in Autism Spectrum Disorders: Relationship to Neuroinflammation. NeuroMol. Med. 2018, 20, 161–173.
- Faravelli, I.; Bucchia, M.; Rinchetti, P.; Nizzardo, M.; Simone, C.; Frattini, E.; Corti, S. Motor neuron derivation from human embryonic and induced pluripotent stem cells: Experimental approaches and clinical perspectives. Stem Cell Res. Ther. 2014, 5, 1–13.
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502.
- Lai, X.; Wu, X.; Hou, N.; Liu, S.; Li, Q.; Yang, T.; Miao, J.; Dong, Z.; Chen, J.; Li, T. Vitamin A Deficiency Induces Autistic-Like Behaviors in Rats by Regulating the RARβ-CD38-Oxytocin Axis in the Hypothalamus. Mol. Nutr. Food Res. 2018, 62, 1700754.
- Cheng, B.; Zhu, J.; Yang, T.; Guo, M.; Lai, X.; Li, Q.; Chen, J.; Li, T. Vitamin A deficiency increases the risk of gastrointestinal comorbidity and exacerbates core symptoms in children with autism spectrum disorder. Pediatr. Res. 2021, 89, 211–216.
- Xu, X.; Li, C.; Gao, X.; Xia, K.; Guo, H.; Li, Y.; Hao, Z.; Zhang, L.; Gao, D.; Xu, C.; et al. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2017, 28, 48–68.
- Khatri, N.; Man, H.-Y. The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front. Mol. Neurosci. 2019, 12, 109.
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3036–3051.
- Ryu, J.; Bar-Shalita, T.; Granovsky, Y.; Weissman-Fogel, I.; Torres, E. Personalized Biometrics of Physical Pain Agree with Psychophysics by Participants with Sensory over Responsivity. J. Pers. Med. 2021, 11, 93.