| Photocatalyst |
Light Source |
Reaction Conditions |
Photocatalytic Performance
(µmolg−1h−1) |
Ref. |
| Au-Ag/TiO2 |
250 W Hg lamp |
Catalyst (14 mg), 20 vol % methanol in water |
12,820 |
[63] |
| Ni-Cd/CdS |
300 W Xe lamp (λ > 410 nm) |
Catalyst (20 mg), Na2S (0.1 M), Na2SO3 (0.1 M) |
11,570 |
[64] |
| FeCu(1:1)@C/g-C3N4 |
300 W Xe lamp |
Catalyst (10 mg), 15 % TEOA in 100 mL water |
722 |
[65] |
| Ni2P/ZnIn2S4 |
300 W Xe lamp (λ > 400 nm |
Catalyst (50 mg), 10 vol % lactic acid in 100 mL water |
2066 |
[66] |
| Cu-nanodiamond |
300 W Xe lamp |
Catalyst (100 mg), 20 vol % ethanol |
1597 |
[67] |
| C3N4-MoS2 nanocomposite |
400 W Xe lamp |
Catalyst (3 mg), 20 vol % TEOA in 40 mL water |
12,778 |
[68] |
3. COFs-Based Hybrids for Photocatalytic CO2 Conversion
CO2 reduction is widely regarded as a technology essential for reducing emissions and providing an alternative means to fossil fuel-based feedstocks, allowing for the sustainable growth and development of human society, using renewable solar energy. An ideal catalyst for CO2 should demonstrate a stable chemical structure, with a band gap suitable for efficiently harvesting visible light, an efficient electron transport, while being strongly absorbing and highly activating. To this end, covalent organic frameworks (COFs) prove an ideal candidate. They represent a class of crystalline polymers, which consist of strong covalently bonded, and highly conjugated bonding systems, with a repeating lattice structure resulting in ordered micropores. The conjugated system promotes an excellent electron mobility, and the covalently bonded system promotes a high thermal and photo-stability. To address the shortfalls of these materials, such as a rapid electron/hole recombination, a variety of inorganic dopants may be introduced, through general wetness impregnation or into vacant coordination sites included in the COF periodic structure. These dopants can serve as active sites, enhancing photocatalytic activity, as well as uptake. Table 5 serves as a summary for synthetic conditions, with Table 6 demonstrating catalytic activity, from the most active to the least active of discussed examples.
Table 5. Synthetic protocols and novelty aspects of hybrid COF-based frameworks explored for CO2 reduction.
| COFs |
Synthesis |
Novelty |
Ref. |
| Co-CTF-1 |
Aerobic acid catalyzed trimerization of terephthalonitrile, with wetness impregnation of Co |
2D-layered COF porous structure with triazine units promoting CO2 adsorption and accommodation |
[69] |
| [Ni(bipy)3]2+—PI-COF |
Anaerobic solvothermal imide condensation between pyromellitic dianhydride and amine-terminated building units. |
Hexagonal porous polyimide COF assisting in CO2 photoreduction |
[70] |
| N3-COF, ACOF-1 |
Anaerobic solvothermal condensation between triformylbenzene and hydrazine hydrate (ACOF-1), and 2,4,6-tris(4-bromophenyl)-1,3,5-triazine with N-formylpiperidine (N3COF) |
Coplanar azine and phenyl rings facilitating the conjugation effect |
[71] |
| Re-bpy-sp2c-COF |
Knoevenagel condensation of 1,3,6,8-tetrakis(4-formylphenyl)pyrene and 5,5′-bis(cyanomethyl)-2,2′-bipyridine. Post synthetic ligation of [Re(CO)5Cl]. |
Porous crystalline COF with bipyridine units ligated to Re complex, forming fully π-conjugated backbone |
[11] |
| COF-367-CoII/III |
Low-pressure solvothermal (50 mTorr). Schiff base condensation between CoII/III-TAPP (5,10,15,20-Tetrakis(4-aminophenyl)porphyrin and 4, 4′-biphenyldicarboxaldehyde. |
CoII and CoIII centered in porphyrin-based COF |
[72] |
| Ru-TpPa-1 |
Anaerobic solvothermal Schiff base condensation between 1,3,5-triformylphloro-glucinol and p-phenylenediamine. RuCl3 added post-synthetically through incipient wetness, followed by reduction in H2/N2. |
Ketoamine-based COF loaded with Ru NPs enhancing the light absorption and charge separation |
[73] |
| Z-Scheme SnS2/S-CTF |
Anaerobic solvothermal nucleophilic substitution between trithiocyanuric acid and cyanuric chloride. SnS2 nanoparticles grown on framework through addition of SnCl2 and heating under autogenous pressure with EtOH. |
Flower-like surface made of vertically aligned nanosheets in which SnS2 nanocrystals are attached with S-CTF framework |
[74] |
| Re-TpBpy |
Anaerobic solvothermal Schiff base condensation between 1,3,5-tri- formylphloroglucinol and 2,2′-bipyridine-5,5′-diamine. Post synthetic ligation of [Re(CO)5Cl]. |
Bipyridine units in COFs facilitating chelation with Re complex creating active sites on the porous walls |
[75] |
| COF-367-Co Nanosheets |
Solvothermal modulated imine exchange, between (5,10,15,20-Tetrakis(4-aminophenyl)porphyrin, 2,4,6-trimethylbenzaldehyde, and 4,4′-biphenyldialdehyde. |
Ultrathin 2D imine-based Co-COF nanosheet structure with large surface area |
[76] |
| DQTP- TMI |
Solvothermal Schiff base condensation between 2,6-diaminoanthraquinone and 1,3,5-triformylphloroglucinol. Transition metal ions (TMIs) Co, Ni and Zn added post-synthetically via hydrothermal treatment. |
2D anthraquinone based COF with conjugation and porosity assisting electron movement and CO2 adsorption |
[77] |
Table 6. COF-based hybrid materials for photocatalytic CO2 reduction.
| COFs |
Type |
Light Source |
Reaction Conditions |
Photocatalytic Performance
(μmol h−1 g−1) |
Ref. |
COF-367-Co
Nanosheets |
Bipyridyl-imine-linked porphyrinic |
300 W Xe
(λ > 420 nm) |
Catalyst (5 mg)
20 mL KOH (aq) (0.1 M)
19 mg [Ru(bpy)3]Cl2.6H2O
352 mg ascorbic acid
1 atm CO2, 25 °C |
Nanosheet:
10,162 (CO)
2875 (H2)
Bulk:
124 (CO) |
[76] |
| Re-bpy-sp2c-COF |
2D sp2c bipyridyl |
300 W Xe
(λ > 420 nm) |
Catalyst (1 mg)
5 mL, 30:1
MeCN:TEOA
1 atm CO2 |
1040 (CO) |
[11] |
| DQTP- TMI |
2D anthraquinone |
300 W Xe
(λ > 420 nm) |
Catalyst (20 mg)
50 mL, 4:1
MeCN:TEOA
22.5 mg Ru(bpy)3Cl2.H2O
1 atm CO2, 25 °C |
TMI = Co: 1022 (CO) |
[77] |
| [Ni(bipy)3]2+—PI-COF |
Covalent Triazine |
300 W Xe
(λ > 420 nm) |
Catalyst (10 mg)
Ni(ClO4)2.6H2O (2 mg)
2,2′-bipyridyl (15 mg)
5 mL, 3:1:1
MeCN: H2O:TEOA
1 atm CO2 |
483 (CO) |
[70] |
| Re-TpBpy |
2D bipyridyl-linked |
200 W Xe
(λ > 390 nm) |
Catalyst (15 mg)
11.8 mL MeCN/H2O (10/1.8)
0.1 M TEOA
1 atm CO2, 25 °C
Purpose-built reactor |
291.7 (CO) |
[75] |
| Z-Scheme SnS2/S-CTF |
Covalent Triazine |
300 W Xe
(λ > 420 nm) |
Catalyst (20 mg),
10 mL, 4:1
H2O:TEOA
1 atm CO2, 25 °C |
123.6 (CO)
43.4 (CH4) |
[74] |
| Ru-TpPa-1 |
Ketoamine-based |
300 W Xe
(λ > 420 nm) |
Catalyst (15 mg)
110 mL, 10:1
MeCN:TEOA
1 atm CO2, |
0 wt % Ru: 32.4 (HCOOH)
1 wt % Ru: 41.8 (HCOOH)
3 wt % Ru: 108.8 (HCOOH)
5 wt % Ru: 61.8 (HCOOH) |
[73] |
| COF-367-CoII/III |
Bipyridyl-imine-linked Porphyrinic |
300 W Xe
(λ > 380 nm) |
Catalyst (10 mg)
22 mL, 10:1
MeCN:TEA
1 atm CO2 |
Co(II)
48.6 (HCOOH), 16.5
(CO), 12.8 (CH4)
Co(III)
93.0 (HCOOH), 0.44
(CO), 10.1 (CH4) |
[72] |
| Co-CTF-T1 |
Covalent Triazine |
300 W Xe
(λ > 420 nm) |
Catalyst (10 mg)
7 mg [Ru(bpy)3]2+
0.9 mL, 1:2:3
TEOA:H2O:MeCN
1 atm CO2 |
48 (CO) |
[69] |
| N3-COF, ACOF-1 |
Covalent Triazine and Azine inked |
500 W Xe
(λ > 420 nm) |
Catalyst (10 mg)
5 mL H2O
4 atm CO2, 80 °C |
N3-COF: 0.57 (MeOH)
ACOF-1: 0.36 (MeOH) |
[71] |
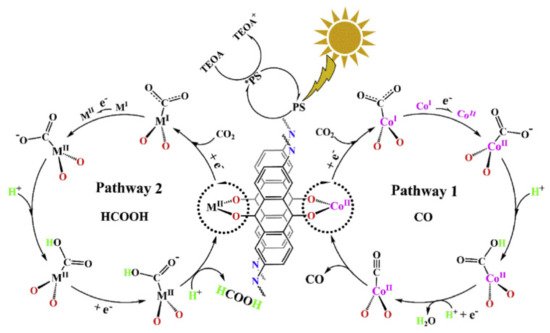
Metal dopant choice was found to strongly influence CO
2 reduction pathways, and the subsequent catalytic activity, in conjunction with the band gap choice of the COF, and the experimental conditions, as highlighted in
Figure 8 [
77].
Figure 8. Proposed CO
2 reduction mechanism over DQTP- TMI COF, highlighting pathways to CO and HCOOH. Reproduced with permission from reference [
77]. Copyright 2019, Elsevier.
Formic acid has been thought to be a suitable hydrogen storage medium, with the ability to decompose to CO
2 and H
2, with a high volumetric capacity (53 g H
2 L
−1), and low toxicity and flammability under ambient conditions [
78]. CO, CH
3OH, and CH
4 also form important chemical feedstocks for assembly into larger hydrocarbons. CO is part of syngas, which can be utilized in the Fischer–Tropsch reaction to form longer hydrocarbons, for use in carbon-neutral fuels [
79]. CH
3OH can be converted into olefins, via the methanol-to-olefin process (MTO), forming the basis for basic chemical feedstocks, or polymer manufacture [
80]. Methane is already widely utilized as natural gas and may be burned in natural gas plants to generate energy or can be converted to syngas for longer chain hydrocarbon synthesis.
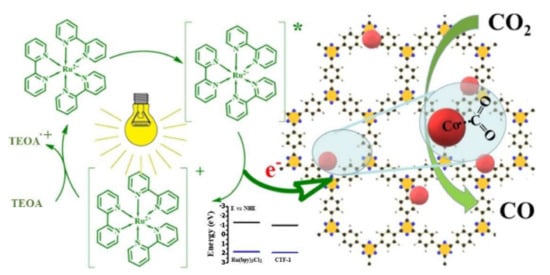
The use of a photosensitizer and a sacrificial electron donor have been found essential for effective photocatalysis. In the work of Fu et al., they demonstrate that undoped ACOF-1 and a triazine framework (based on 2,4,6-tris(4-bromophenyl)-1,3,5-triazine) yield a low activity when tested at 4 atm of CO
2, at 80 °C, with 8.6 μmolg
−1 of MeOH for ACOF-1, and 13.7 μmolg
−1 for N
3-COF [
71]. When compared to other materials examined in
Table 6, which utilize metal dopants, photosensitizers, and sacrificial electron donors, they demonstrate the lowest activity. However, a key difference is that the N3-COF demonstrates a higher activity than ACOF-1, which instead consists of an azine linked benzene structure. Triazine-based COFs tend to give a higher performance than non-triazine systems as they have a higher affinity for negative charge when conducting electrons [
69,
70,
71,
81]. However, one shortfall of organic semiconductors is a rapid electron-hole recombination [
74]. A photosensitizer prevents this, facilitating a higher catalyst efficiency, and may be used as a homogeneous co-catalyst, or itself be incorporated into the COF structure post-synthetically.
For example, Bi et al. report that the inclusion of Co
2+ into a triazine based CTF-1 causes a 44-fold enhancement (48 μmolg
−1 CO) in catalytic activity over that of the pristine framework (1.1 μmolg
−1 CO) [
69]. Notably, when comparing to the work of Fu et al., both a photosensitizer, [Ru(bpy)
3]
2+, and a sacrificial donor, triethanolamine (TEOA) are used. As
Figure 9 outlines, the photosensitizer is excited by incoming light, and separates the electron/hole pair by transferring the generated electron to the COF, which reduces CO
2. TEOA then sacrifices an electron, quenching the photosensitizer. Cobalt was thought to enhance this mechanism through providing single atom active sites, which can readily adsorb CO
2, as well as restraining electron/hole recombination rate within the COF. CO
2 adsorption was found to be higher for the impregnated Co-CTF (9.76 cmg
−1) compared to the pristine (7.97 cmg
−1) and normalized to surface area. Zeta potential findings indicated that Co provides suitable Lewis acid sites for the coordination of CO
2. Photoluminescence spectroscopy found that the Co-CTF demonstrated a longer lifetime than that of the pristine, demonstrating the longer-lived charge carriers in the Co-CTF.
Figure 9. Interaction of a [Ru(bpy)
3]
2+ with sacrificial donor TEOA and Co-CTF framework. Reproduced with permission from reference [
69]. Copyright 2019, Wiley-VCH.
Researchers have further experimented with metal dopant inclusion into COF structures by integrating the photosensitizer as part of a COF structure. For example, the work of Fu et al. demonstrates a 2D sp
2c bipyridyl based COF, synthesized via a Knoevenagel condensation, in which a photosensitizer like-complex of Re(bpy)(CO)
2Cl has been incorporated into the structure of the framework, through the bipyridyl nitrogen binding sites [
11]. Successful chelation was confirmed via XPS, where the Re doped structure gave a similar XPS trace as free [Re(bpy)(CO)
3Cl], demonstrating the chelation of Re. This was also supported by FTIR observations, where in addition to the triazine vibration at 2217 cm
−1, CO stretching bands of 1900, 1917, and 2024 cm
−1 was observed. By including Re in the structure, a CO yield of 1040 μmolg
−1 was obtained, whereas without Re, only traces of CO was observed, demonstrating the need for a photosensitizer to facilitate electron/hole generation. The interaction of Re with the framework was rationalized through the following observations. UV-Vis indicated a red shifting of the absorption edge of the material from 589 nm to 694 nm, demonstrating a slight alteration of bandgap on Re addition. With the addition of Re, the BET surface area of the pristine, 432 m
2g
−1 is lowered to 323 m
2g
−1, although the CO
2 sorption capability of the material is improved, with the Re-COF adsorbing 1.7 mmolg
−1 of CO
2 at 273 K, with a high isosteric heat of adsorption, 31 kJmol
−1. The photocurrent response of the COF is increased with the inclusion of Re, indicating a higher current density being generated, and the Nyquist plots of the Re-doped COF show smaller arc radii than the undoped material, signifying that the addition of Re aids in charge separation and transfer.
Guo et al. demonstrate that this effect is related to the percentage loading of metal, when doping ruthenium NPs into TpPa-1, where the 0 wt % loading demonstrated the lowest yield of CO at 32.4 μmolg
−1h
−1, with 1, 3, and 5 wt % yielding 41.8, 108.8, 61.8 μmolg
−1h
−1, respectively [
73]. TpPa-1 was synthesized solvothermally under anaerobic conditions achieved through the evacuation and sealing of reactants in a glass ampoule. Ru was then introduced to the material via wetness impregnation, before being dried and reduced at 280 °C for 6 h. The known thermal stability of most COF structures at these temperatures permits elevated temperatures for post-synthetic treatment, although some evidence suggests that thermal treatment of 2D COFs can alter the periodic structure, which is not detectable via TGA [
82]. The resulting Ru loading was determined via ICP-OES, with the nanoparticle morphology confirmed via HR-TEM, with [1 0 0] and [2 1 0] lattice planes evident, characteristic of metallic Ru. Notably, no Bragg peaks for Ru were observed via PXRD, indicating a high dispersal dispersion of Ru throughout the framework, also verified through EDX analysis. XPS analysis indicated a shift for the Ru 3p1/2 and 3p3/2 signals (484.3 and 462.1 eV, respectively) vs. the RuCl
3 (485.6 and 463.4 eV) standard, indicating the complete reduction of Ru
3+ to Ru
0. Photoluminescence spectroscopy indicated that the shortest lifetime was observed with the 3 wt % loaded sample, and transient photocurrent responses indicated the highest charge density on irradiation with light. Both results suggest that 3 wt % is the optimal loading for charge separation. Nyquist plots also yielded the smallest arc radii for the 3 wt % sample. It was thought that by increasing the loading beyond 3 wt %, Ru clusters are being formed which begin to facilitate charge recombination, thus lowering the efficiency of the material.
Chen et al. demonstrated that the dopant species does not necessarily have to be a precious metal [
70]. Their work focused on comparing polyimide linked COFs, with a [Ni(bpy)
3]
2+ photosensitizer. When comparing tris(4-aminophenyl)amine (TAPA), 1,3,5-tris(4-aminophenyl) benzene (TAPB), or 1,3,5-tris(4-aminophenyl)triazine (TAPT) structures connected via polyimide linkages, the azine-based COF gave the highest performance of 483 μmolg
−1h
−1, whereas only trace activity was observed for the other tested frameworks. The imide condensation was performed solvothermally, with the reaction mixture degassed via pump-thaw through flash freezing in liquid nitrogen, before being sealed and heated at 200 °C for 5 days. All materials indicated a successful condensation, with characteristic stretching bands of 1720–1725 cm
−1 indicating the 5-member imide ring formation, and an imide carbonyl signal observed at 165 ppm via
13C NMR. All materials matched with their simulated PXRD patterns, with a weakening of the [1 1 0] peak on Ni addition, suggesting an increase of disorder for this lattice plane. The surface area values for the materials were within expected ranges, with 475, 1175, and 825 m
2g
−1 for TAPA, TAPB, and TAPT, respectively, with all materials exhibiting a pore size of 1.5–3.5 nm, modelled using nonlocal density functional theory (NLDFT). The TAPT catalyst demonstrated the highest isosteric heat of absorption adsorption of CO
2 (29.76 kJ mol
−1), indicating the attraction between the triazine functionality and CO
2, and XPS data demonstrated that the doped [Ni(bpy)
3]
2+ was encapsulated in the pore system, based on an intensification of the Ni 2p signal through argon ion bombardment. When examined electrochemically, photocurrent density was found to be the highest for the TAPT material, mirroring catalytic results. To explore this further, EPR experiments were carried out, comparing a sample before and after irradiation. The shift in electron density to the triazine ring demonstrated the capability for the functional group to hold negative charge, facilitating charge separation compared to the other conjugated systems examined in this work.
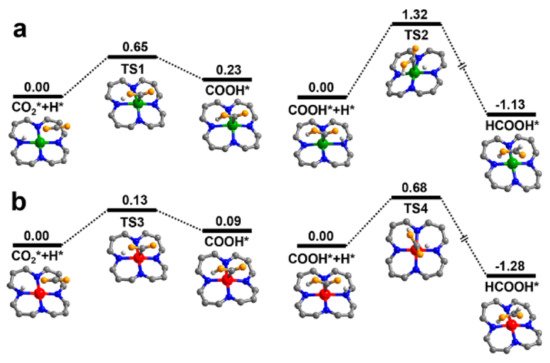
The electronic environment of the dopant species was also found to be crucial for product selectivity, as found by Gong et al. [
72]. When examining Co-COF-367, a porphyrinic COF, it was found that Co
II-COF-367 has a product profile of 48.6, 16.5, and 12.8 μmolg
−1h
−1 of formic acid, CO and methane, whereas Co
III-COF-367 yields 93.0, 0.44, and 10.1 μmolg
−1h
−1 of the respective products. Using
13C labelled CO
2 as a feedstock, the researchers confirmed that all products are a result of photocatalytic reduction. EPR measurements indicated a strong signal for Co
II at g = 2.30 while the material was measured in the dark where the intensity decreased on irradiation with light. This was thought to signify the transformation of Co
II to Co
I. The density functional theory (DFT)-based calculations indicate that the presence of Co
II is beneficial for forming formic acid, but prevents any further conversion, due to a high barrier energy for transformation into CO or CH
4. Co
III dopants demonstrate a lower barrier energy, vindicating the experimental observations as shown in
Figure 10. Photoelectrical experiments indicate that Co
III-COF-367 has a higher charge separation efficiency than the Co
II structure, demonstrated by a longer photoluminescent activity with photoluminescence spectroscopy, as well as a higher photocurrent response.
Figure 10. Calculated potential energy profile of CO
2 to HCOOH, over (
a) COF-367-Co
II, and (
b) COF-367-Co
III. Co
II, Co
III, C, N, O, and H atoms are shown as green, red, gray, blue, orange, and light gray, respectively. Reproduced with permission from reference [
72]. Copyright 2020, American Chemical Society.
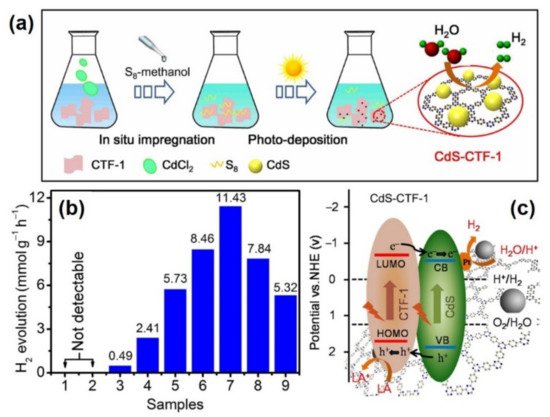
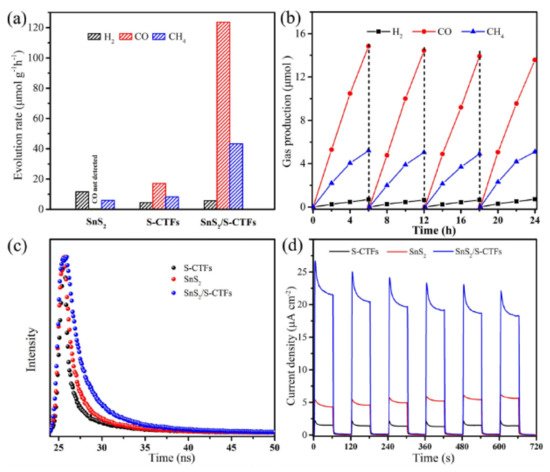
Guo et al. demonstrate that charge separation efficiency can be achieved through heterojunction structures, by doping a sulfur-bridged triazine COF with SnS
2 [
74]. The hybrid material demonstrated a greatly enhanced performance for CO
2 photoreduction, demonstrating average evolution rates of CO and CH
4 up to 123.6 and 43.4 μmol h
−1 g
−1 respectively, when irradiated under visible (420 nm) light, as demonstrated in
Figure 11a. By comparison, SnS
2 evolved only CH
4 at a rate of 5.9 μmol h
−1 g
−1, whereas S-CTF evolved both CO and CH
4 at rates of 17.1 and 8.3 μmol h
−1 g
−1, respectively. The hybrid material also demonstrated significant photostability, with performance showing little attenuation after four 6-h cycles, shown in
Figure 11b. By designing a Z-scheme heterojunction, catalysis shows that recombination of electron/hole pairs are hindered, increasing catalytic efficiency compared to the isolated materials. Photoluminescence spectroscopy demonstrated that recombination was significantly quenched for SnS
2/S-CTF. The average PL lifetime demonstrated by the hybrid reached 7.18 ns, compared to 5.60 ns and 4.28 ns for SnS
2 and S-CTF (
Figure 11c). The S-CTF was synthesized solvothermally under anaerobic conditions via the nucleophilic substitution of cyanuric chloride and trithiocyanuric acid. SnS
2 was doped in the form of NPs, which were formed in-situ by immersion of the S-CTFs in a SnCl
2 ethanol solution at 180 °C. SEM imaging of the pristine S-CTF showed the formation of 400 nm smooth spheres. The introduction of SnCl
2 triggered the formation of vertical platelets on the surface of the spheres, of which high resolution TEM indicates are composed of <5 nm nanocrystals. The lattice fringes of said crystals were found to correspond with the (1 0 1) and (1 0 0) planes of crystalline SnS
2, supporting PXRD findings demonstrating diffraction planes of both S-CTF and SnS
2 in the hybrid material. To further support the formation of a heterojunction, XPS was used to examine the nature of the binding energies of surface N species. Compared to the undoped S-CTF counterpart, the doped material exhibited an increase in N species binding energy by 0.5 eV. This was interpreted as evidence for an electron transfer from the framework to the inorganic nanoparticle. This resulted in a higher observed transient photocurrent response for the hybrid material (
Figure 11d).
Figure 11. (
a) Average gas production rates of SnS
2, S-CTFs, and SnS
2/S-CTFs. (
b) Photostability of SnS
2/S-CTFs for repeat CO
2 reduction cycling. (
c) Time resolved PL decay curves of S-CTFs, SnS
2, and SnS
2/S-CTFs. (
d) Transient photocurrent responses under λ > 420 nm irradiation. Reproduced with permission from reference [
74]. Copyright 2020, Wiley-VCH.
The morphology of COF nanostructures also plays an important role in affecting the photocatalytic performance. For the materials considered in this review in
Table 5, all 2D structures have been found to have some of the highest performances. The 2D structures have more accessible pores than 3D bulk crystalline structures, as well as a better conductivity, which facilitates photoelectric efficiency [
11,
75,
76,
77]. As previously discussed, Fu et al. demonstrate a Re-functionalized 2D sp
2c bipyridyl based COF, with a performance of 1040 μmolg
−1h
−1 [
11]. The work by Li et al. demonstrates a similarly Re-functionalized 2D “TpBpy” COF, with a performance of 291 μmolg
−1h
−1 [
75]. When compared to a homogeneous Re-bpy catalyst, the Re-COF gave a higher performance over time due to the porosity of the structure ensuring a higher contact time between the Re active site, compared to the exposure of the homogeneous catalysts which is based on CO
2 diffusion through the reaction medium. According to CO
2 sorption experiments, Re-TpBpy demonstrated an adsorption volume of 44 cm
3g
−1, whereas the adsorption volume was near 0 for the Re-Bpy catalyst. XPS validated the electronic interaction of the Re functionalization on the framework, with a shift of the pyridinic N 1s signal from 398.9 to 400.46 eV on Re addition. This was associated with a decrease of electron density as Re is complexed.
Restricting morphology further to 2D nanosheets has been found to significantly impact the catalytic performance. Liu et al. demonstrate that by fabricating Co-COF-367 nanosheets using a modulated imine-exchange bottom-up synthesis strategy, catalytic activities of up to 10,162 and 2875 μmolg
−1h
−1 for CO and H
2 respectively, were evidenced [
76]. An analogous bulk Co-COF-367 demonstrated an activity of only 124 μmolg
−1h
−1. However, it is notable that in the absence of the [Ru(bpy)
3]Cl
2 photosensitizer or the ascorbic acid sacrificial agent, only trace activity was observed. Synthesis of the nanosheets used H
2TAPP alongside 4,4′-biphenyldialdehyde (BPDA) as building units, with 2,4,6-trimethylbenzaldehyde (TBA) functioning as the modulator. By using 4-bromo-2,6-dimethylbenzaldehyde in place of the modulator, nanosheets were fabricated which bromine “marked” lateral planes, observable via elemental mapping, in
Figure 12, which vindicated the postulated effect of the modulating agent. For purification, bulk COF impurities were separated from the final product through suspension and centrifugation at 2000 rpm, yielding a colloidal dispersion of the nanosheets. PXRD found that the nanosheets were not crystalline enough to yield meaningful diffraction peaks. As a result, scanning tunneling microscope (STM) was used to reveal the periodic pore lattice of the structure, giving lattice parameters of a = b = 2.7 nm, α = 90, which matched with the simulated structure. Sharp selected area electron diffraction (SAED) was used to highlight the ordered tetragonal structure of these sheets, to verify the lattice order of the sheets despite the low crystallinity. Bulk frameworks did yield a higher BET surface area, 467 m
2g
−1 of the bulk, vs. the 106 m
2g
−1 of the nanosheets.
Figure 12. (
a) TEM image (
b) HAADF-STEM image and (
c) elemental mapping of COF-367 nanosheets, modulated using 4-bromo-2,6- dimethyl benzaldehyde, with the lateral plane demonstrating a high concentration of the modulator. Adapted with permission from reference [
76]. Copyright 2019, American Chemical Society.
Product modulation can also be achieved within 2D structures. Lu et al. demonstrated this with a 2D 2,6-diaminoanthraquinone-based COF, functionalized with Co, Ni and Zn [
77]. The type of dopant-controlled CO production, with a 1002 μmolg
−1h
−1 production of CO with Co, and a 152.5 μmolg
−1h
−1 yield of formic acid over the Zn based material. The framework was synthesized via a Schiff-base condensation, and then metallated post-synthetically through refluxing with the metal salt in solution. XPS was used to verify the presence of metals, with the following environments found; Co 2p
3/2 = 786.2 eV, as evidence for Co
II, Ni 2p
3/2 = 855.6 eV for Ni
II, and Zn 2p
3/2 = 1022.2 eV for Zn
II. The Co energy is red shifted, which was taken as evidence for a Co-O bond with neighboring carbonyls, which donate electron density. Obvious bonding signals were not found for Ni and Zn dopants. Interaction with framework carbonyls was demonstrated further, through instead using a 2,6-diaminoanthracene group in place of the 2,6-diaminoanthraquinone. This demonstrated a significantly lower activity for Co dopants, but affected Ni and Zn dopants much less. These findings, amongst others, were used to elucidate a two-path mechanism for photoreduction over these systems, which was found influenced by metal type. In addition to this, Lu et al. also noted the importance of the sacrificial donor type. It was found that TEOA favors gaseous products CO, whereas TEA and TIPA favor formic acid. It is still a matter of discussion as to why this is the case.
Table 7 summarizes the structure-property relations of the catalytic systems discussed in this section for CO
2 reduction reaction. The catalytic active sites and their topology has been correlated with their photocatalytic performance. The ultrathin imine-based Co-COF nanosheet framework with large surface area values displayed the most active photocatalytic reduction of CO
2 to CO (10162 µmolh
−1g
−1). The Re-bpy-COF with bipyridine units exhibited second highest reaction rate due to the extended conjugation offered by the Re complex. According to the recently reported studies, the state-of-the-art photocatalysts for CO
2 reduction also displays comparable activity to the COF materials [
83,
84].
Table 8 summarizes some of the photoactive materials including Ag-Cr/Ga
2O
3 (CO, 480 µmolg
−1h
−1) [
85], Cu@V-TiO
2/PU (CH
4, 933 µmolg
−1h
−1) [
86], ZnO nanosheets (CO, 406.77 µmolg
−1h
−1) [
87], Rh-Au-SrTiO
3 (CO, 369 µmolg
−1h
−1) [
88], Mo-doped g-C
3N
4 (CO, 887 µmolg
−1h
−1) [
89] and Nb-TiO
2/g-C
3N
4 (CH
4, 562 µmolg
−1h
−1) [
90].
Table 7. Structure–activity correlations for enhancing the photocatalytic performance in CO2 reduction reaction.
| COFs |
Active Sites |
Topology |
Photocatalytic Performance (µmolh−1g−1) |
Ref. |
| Co-CTF-1 |
COF and Ru(bpy)32+: photosensitizer
Co: redox active sites |
2D-layered COF porous structure with triazine units promoting CO2 adsorption and accommodation |
48 (CO) |
[69] |
| [Ni(bipy)3]2+—PI-COF |
COF: photosensitizer
Ni: CO2 activation |
Hexagonal porous polyimide COF assisting in CO2 photoreduction |
483 (CO) |
[70] |
| N3-COF, ACOF-1 |
COF: photosensitizer and redox active site |
Coplanar azine and phenyl rings facilitating the conjugation effect |
N3-COF: 0.57 (MeOH)
ACOF-1: 0.36 (MeOH) |
[71] |
| Re-bpy-sp2c-COF |
COF: photosensitizer
Re complex: electron mediator |
Porous crystalline COF with bipyridine units ligated to Re complex, forming fully π-conjugated backbone |
1040 (CO) |
[11] |
| COF-367-CoII/III |
COF: photosensitizer
Co: redox active site |
CoII and CoIII centered in porphyrin-based COF |
Co(II)
48.6 (HCOOH), 16.5 (CO), 12.8 (CH4)
Co(III)
93.0 (HCOOH), 0.44 (CO), 10.1 (CH4) |
[72] |
| Ru-TpPa-1 |
COF: photosensitizer
Ru NPs: reductant |
Ketoamine-based COF loaded with Ru NPs enhancing the light absorption and charge separation |
0 wt % Ru: 32.4 (HCOOH)
1 wt % Ru: 41.8 (HCOOH)
3 wt % Ru: 108.8 (HCOOH)
5 wt % Ru: 61.8 (HCOOH) |
[73] |
| Z-Scheme SnS2/S-CTF |
SnS2 and COF: photosensitizer
COF: CO2 adsorption and activation |
Flower-like surface made of vertically aligned nanosheets in which SnS2 nanocrystals are attached with S-CTF framework |
123.6 (CO)
43.4 (CH4) |
[74] |
| Re-TpBpy |
COF: photosensitizer
Re complex: catalytic active site |
Bipyridine units in COFs facilitating chelation with Re complex creating active sites on the porous walls |
291.7 (CO) |
[75] |
| COF-367-Co Nanosheets |
Ru complex: photosensitizer
Co-COF: active site |
Ultrathin 2D imine-based Co-COF nanosheet structure with large surface area |
Nanosheet:
10162 (CO)
2875 (H2)
Bulk:
124 (CO) |
[76] |
| DQTP- TMI |
Ru complex: photosensitizer
Co, Ni, Zn-COF: redox active site for CO2 adsorption and reduction |
2D anthraquinone based COF with conjugation and porosity assisting electron movement and CO2 adsorption |
TMI = Co: 1022 (CO) |
[77] |
Table 8. State-of-the-art photocatalysts for CO2 reduction.
| Photocatalyst |
Light Source |
Reaction Conditions |
Photocatalytic Performance (µmolh−1g−1) |
Ref |
| Ag-Cr/Ga2O3 |
400 W Hg lamp (λ > 420 nm) |
Catalyst (0.5 g), NaHCO3 (0.1 M), CO2 (30 mLmin−1) |
480 (CO) |
[85] |
| Cu@V-TiO2/PU |
Two white light bulbs |
CO2 (50 mLmin−1) passing through water at 303 K |
933 (CH4) |
[86] |
| ZnO nanosheets |
125 W Hg lamp |
Catalyst (50 mg), 0.5 mL deionized water at 473 K, CO2 (1 atm) |
406.77 (CO)
20.16 (CH4) |
[87] |
| Rh-Au-SrTiO3 |
500 W Xe lamp
(λ > 400 nm) |
Catalyst (75 mg), 3 mL deionized water and CO2 (70 kPa) |
369 (CO) |
[88] |
| Mo-doped g-C3N4 |
300 W Hg lamp |
Catalyst (100 mg), 5.0 g deionized water at 303 K and CO2 (110 KPa) |
887 (CO)
123 (CH4) |
[89] |
| Nb-TiO2/g-C3N4 |
Two 30 W white bulbs |
Catalyst (100 mg), CO2 (20 mL min−1) passing through water at 303 K |
562 (CH4)
420 (CO)
698 (HCOOH) |
[90] |