The kidney is one of the main organs affected by the autoimmune disease systemic lupus erythematosus (SLE). Lupus nephritis (LN) concerns 30–60% of adult SLE patients and it is significantly associated with an increase in the morbidity and mortality. The definitive diagnosis of LN can only be achieved by histological analysis of renal biopsies, but the invasiveness of this technique is an obstacle for early diagnosis of renal involvement and a proper follow-up of LN patients under treatment. The use of urine for the discovery of non-invasive biomarkers for renal disease in SLE patients is an attractive alternative to repeated renal biopsies, as several studies have described surrogate urinary cells or analytes reflecting the inflammatory state of the kidney, and/or the severity of the disease.
- lupus nephritis
- urine biomarkers
- non-invasive diagnosis
- immune effector
1. Introduction
Kidney biopsy is the gold standard for establishing tissue diagnosis, prognosis and treatment in LN. However, it is a costly and risky procedure, making it unsuitable for the early detection of renal pathology or to monitor the response to treatment. Nowadays, standard laboratory markers of kidney involvement have been applied in the monitoring of LN activity in daily clinical routines [4]. Nonetheless, these analytical parameters lack sensitivity and specificity to reflect the real-time renal immunopathological activity and chronic tissue damage [5]. Current treatment of LN patients involves immunosuppressive therapy and glucocorticoids. However, they are neither uniformly effective nor specific of the disease, and they have shown side effects [6]. Therefore, novel biomarkers able to discriminate lupus renal activity and its severity, predict renal flares, and monitor treatment response and disease progress are clearly necessary.
In contrast to other biological sample sources, urine sampling is non-invasive, allowing frequent monitoring, and can be self-collected, transported, and stored easily. Furthermore, urinary biomarkers seem to be more promising than serum markers in the study of LN, given that they derive from tissues of the urinary system [7], so that they can reflect its current pathological status [8]. Thus, urine is an attractive source for finding potential biomarkers in the study of LN.2. Autoantibodies
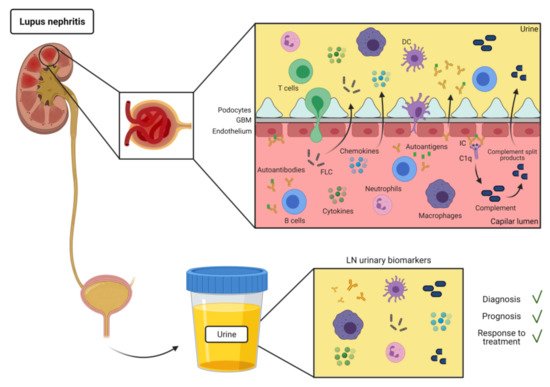
SLE is an autoimmune disease characterized by production of autoantibodies against self-molecules present in the nucleus, cytoplasm, and cell surface. There is evidence of the presence of IC deposition in renal biopsies of patients with LN [9]. IC formation and deposit in the kidneys are most likely involved in the mechanism for urinary excretion of autoantibodies in SLE (Figure 1). However, there have been few reports of specific autoantibodies in the urine of SLE patients (Table 1).
Autoantibodies anti-Sm, anti-RNP, anti-SSA and anti-dsDNA have been found in the urine of SLE patients, many of them with signs of abnormal renal function such as proteinuria [10][11][12]. Autoantibodies anti-SSB, anti-ribosomal P and anti-RNAPI have also been observed in urine of SLE patients. In addition, the relative concentration of these urinary autoantibodies correlated with disease activity [13].
Apart from anti-nuclear autoantibodies, anti-IFNa have been detected in urine of SLE patients. Specifically, autoantibodies against both IFNα1 and IFNα6 were observed in urine but not in serum of SLE patients, suggesting that local immune responses in kidney would be distinct from those in serum [14].
| Urinary Biomarker Class |
Diagnostic Value | Prognostic Utility | Response to Treatment |
|---|---|---|---|
| Autoantibodies | Anti-RNAPI, anti-dsDNA, anti-La, and anti-ribosomal P, levels correlated with disease activity [13] | ||
| FLC | FLC discriminate patients with severe forms of LN [15][16] | FLC increase before the onset of acute SLE relapses and reach normal values after remission [17][18] | λ and κ FLC decrease after treatment [16] |
| Complement components | C3d levels correlate with SLEDAI discriminate between active LN and inactive LN or non-renal SLE [32][37][38] | C3d decreased levels can predict treatment response at 6 months and non-response or flare [38] | C3d levels fall after therapy [38] |
| Soluble immune mediators | IL-6 higher in active LN [15] corroborated by renal biopsy [42] | No differences between active or inactive LN [43]. | Decreased significantly after treatment [41] |
| MCP-1 correlates with LN activity [44]. Higher in patients with inactive LN (Meta-analysis) [45]. Increased also in serum of SLE patients [47] MCP-1, KIM-1, and NGAL higher in patients with active LN [46] |
|||
| IP-10 positively correlated with renal SLEDAI but not significantly higher in LN [49] | |||
| EGF lower in patients with active LN [50] | Decreased overtime in adverse long-term kidney damage [50] | ||
| VCAM-1 higher in active renal disease [51]. Presence of LN, clinical and histological activity indexes severe renal lesions [52][53] VCAM-1 and ALCAM elevated in active LN [54][55] ICAM-1 elevated in SLE patients [56] VCAM-1, cystatin C, and KIM-1 discrimination between proliferative versus membranous LN. Non-specific for SLE [57] NGAL; higher in LN than in non-LN patients [59] |
Increased in active LN [53]. It may indicate patients at increased risk for long-term renal function loss [54]. ALCAM levels correlated positively with activity index [55] |
Effective LN therapy reduced uVCAM-1 levels over the time [53] | |
| Leukocytes | Monocytes/macrophages in proliferative LN [61][62][63] Higher eosinophils numbers in LN [73] CD4+CD25-Foxp3+ T cells in active LN [80] CD4+ and CD8+ T cells in active LN [75][76] Th17 associated with less severe disease [79] pDC and PB/PC in severe LN [81][83] |
Lower numbers of CD8+ T cells in remission [75][76] | |
| Soluble leukocyte markers | sCD163 in active LN [68][70], mostly in proliferative classes [71] sCD11b correlates with histological activity [72] T-bet mRNA in higher in active LN [74][78] |
sCD163 increases from 6 months before flare [69] Higher T-bet mRNA gives higher risk of severe flare [78] |
sCD163 decreases after treatment in drug responders [69][70] sCD11b decreases with clinical remission [72] |
In summary, although there are very few studies reporting the clinical significance of urine autoantibodies, the analysis of their distinct specificities in urine and serum has the potential to become a useful tool for the diagnosis and monitoring of the renal disease activity in SLE patients. Nevertheless, this should be confirmed in larger and serial sampling studies.
3. Free Light Chains
B cell activation has an important role in the pathogenesis of SLE. During the flares, an extensive polyclonal B cell hyperactivity is observed, followed by an exacerbated synthesis of immunoglobulins, made of two heavy chains and two free light chains (FLC): κ and λ [19]. In chronic inflammatory diseases such as SLE, elevated levels of urine FLC can be found as a result of increased production overcoming the renal clearance capacity or renal tubule impairment [20][21].
The increase in FLC in the urine of patients with SLE has been described during active renal disease [22]. Later studies reported a significant increase in urinary FLC 4–8 weeks prior to the onset of acute SLE relapses, suggesting that a time frame may exist between the occurrence of immunopathologic B-cell stimulation and the resultant symptoms and tissue damage mediated by IC-induced acute inflammation [17]. Urinary FLC were increased during active phases of SLE, whereas they reached normal values after remission [18]. Similar to increased levels of soluble interleukin-2 receptors, FLC and the presence of cytokine-like molecules in urine can directly reflect the severity of inflammatory and immunological reactions in patients with LN [15] (Table 1).
A study comparing serum and urinary FLC in SLE patients revealed patients presented high urinary FLC in the absence of detectable serum FLC [23]. Levels of urinary FLC in LN patients with class III/IV were higher than in non-class III/IV LN patients. Levels of FLC in urine showed a strong correlation with proteinuria and plasma cell infiltration of the kidney. Furthermore, both urinary and serum FLC were lower after treatment, thus providing evidence for a possible direct biomarker of renal inflammations and local pathogenesis [16] (Table 1).
FLC play an important role in the renal pathogenesis of SLE. All these studies showed significant findings in the field of urinary FLC as biomarkers for LN. However, urinary FLC are not specific for LN [16]. Therefore, more studies of FLC in urine may help to understand their role in different diseases, and provide a useful parameter to monitor disease progression. Furthermore, larger longitudinal studies are needed to determine the predictive value of urinary FLC as biomarkers of disease activity and relapse, as well as treatment response.4. Complement
The involvement of the complement in the pathogenesis of SLE is well accepted, although its exact role is still not clear. Hereditary deficiencies of complement components are associated with an increased risk for the disease [24]. The deposition in the kidney of IC formed by autoantibodies directed against self-antigens, and the subsequent activation of the classical pathway are considered major mechanisms mediating tissue injury in LN [25]. Moreover, the involvement of the alternative or lectin pathways has also been suggested in several studies [26][27][28][29][30].
It has long been recognized that serum C3 and C4 levels generally are lower in SLE patients [31]. However, serum low complement levels have proven disappointing as disease activity markers in SLE due to their persistency at low or normal levels, independently of disease activity, and their low sensitivity at predicting flares [25]. Thus, urine detection of complement products in urine is still an attractive option.
It has been described that complement components can be found in the urine of LN patients, particularly in patients with active kidney disease. These studies have reported the presence of different complement proteins in the urine of patients with LN [32][33][34][35]. The most studied has been urine levels of fragmented C3d. Detection of urinary excretion of C3d fragments among SLE patients was variable among SLE patients with non-renal manifestations [36]. Other studies found C3d in urine of patients with LN, but also in patients with no evidence of renal disease or proteinuria, suggesting a non-renal origin of C3d. Nevertheless, urine C3d levels were better than plasma C3, C4, C4d, C5b-9, and anti-dsDNA to differentiate acute from chronic LN [32]. C3d were elevated in urine of patients with active LN compared to inactive LN and non-renal SLE. Moreover, urine C3d showed a stronger correlation with the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) than serum C3, C3d, C4, or anti-dsDNA antibodies (Table 1) [37]. Finally, in a longitudinal study, LN patients showed high levels of urinary C3d at onset of the disease and a significant fall after 3 months of treatment. In addition, levels of urinary C3d correlated with SLEDAI, but there was no significant correlation with plasma C3 and C4, suggesting an intra-renal production of C3d (Table 1). Urinary C3d/creatinine values could discriminate between active and inactive nephritis [38].
In summary, complement fragments indicative of complement activation can be found in the urine of LN patients, particularly in patients with active kidney disease. These studies support the idea that urinary C3d levels correlate more tightly than other markers with LN disease activity, so that they can be used as biomarkers to determine response to treatment, identify non-responders or relapses. However, larger studies are required to further validate the use of urinary C3d as a biomarker of LN.5. Soluble Immune Mediators
Most of the soluble immune-related molecules present in urine includes cytokines and chemokines, growth factors and others (Figure 1). They are easy to detect and their increase can be indicative of inflammation [39][40].
Urine IL-6 was described as a potential marker to follow the disease as it was higher in patients with LN class IV. Additionally, IL-6 levels decreased significantly after treatment [41]. In a more recent study, urinary levels of IL-6 were higher in SLE patients with LN, corroborated by biopsy [42]. Levels of urinary IL-6 were able to discriminate between active or inactive LN [43] (Table 1).
Urine levels of monocyte chemoattractant protein 1 (MCP-1) have been proposed to correlate with LN activity [44]. Urinary level of MCP-1 was higher when comparing active LN with inactive LN patients, or with healthy controls. Level of MCP-1 was higher in patients with inactive LN than in healthy controls [45]. Other studies have evaluated the levels of MCP-1 together with other chemokines. Levels of MCP-1, kidney injury molecule (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) were higher in patients with active nephritis, compared to LN patients in remission and normal controls [46]. In a recent study, a significant increase in MCP-1 was detected in serum and urine of all SLE patients [47].
Another study found significant differences between active LN and non-renal SLE for vascular endothelial growth factor (VEGF), and levels of soluble tumor necrosis factor receptor 1 (sTNF-R1) and interferon-inducible protein 10 (IP-10) in urine and serum correlated with SLEDAI scores. sTNF-R1 and VEGF have also been proposed as markers of disease activity in SLE and LN[48]. Urine level of IP-10 showed a non-significant trend to be higher in patients with active LN, even though IP-10 level showed a positive correlation with renal SLEDAI [49].
Urinary Epidermal Growth Factor (EGF) has also been evaluated as a possible LN biomarker (Table 1). Its urinary level was lower in patients with active LN compared to patients with active non-renal SLE, patients with inactive SLE and healthy kidney donors. Follow-up of the patients corroborated that urinary EGF were lower at flare and were decreasing over time in the case of adverse long-term kidney damage [50].
Vascular cell adhesion molecule 1 (VCAM-1) has been widely studied in LN (Table 1). VCAM-1 was significantly higher in urine from patients with active renal disease compared to patients with active non-renal disease [51]. Soluble urine VCAM-1 showed a strong association with the presence of LN, with clinical and histological activity indexes and with more severe renal lesions [52][53]. Urine levels of VCAM-1 and ALCAM were elevated in patients with active LN compared to healthy controls and with quiescent nephritis. A positive correlation was established between urine ALCAM and SLEDAI. ALCAM level was higher in proliferative the classes III and IV. Of both, higher levels of VCAM-1 in urine could be indicative of a long-term renal function loss, while ALCAM could be used as a potential biomarker of kidney disease [54][55].
Another molecule with a relevant role in LN is the intercellular cell adhesion molecule ICAM-1. ICAM-1 level was higher in SLE patients (Table 1). Similarly, ICAM-1 was increased in the blood, but did not allow the discrimination between active and inactive SLE [56]. Study of VCAM-1 along with other urinary biomarkers revealed that values of VCAM-1, cystatin C, and KIM-1 allowed to discriminate between proliferative versus membranous LN. However, these markers are not specific for SLE [57].
Finally, there are other immune-related enzymes or molecules present in urine from SLE patients that could be used as biomarkers. NGAL appeared increased in urine after renal injury [58] and its urinary levels in LN patients were significantly higher than in non-LN patients [59].
In conclusion, there are several cytokines, chemokines and other soluble immune mediators present in urine that could be used as biomarkers of kidney damage in SLE. However, we still do not have a definitive panel of molecules allowing a precise diagnosis of renal damage in SLE or to differentiate the stages of renal disease.6. Cell-Associated Biomarkers
Urine leukocytes and erythrocytes are often present in the urine of LN patients (Figure 1). Urine cells can be collected by urine centrifugation and analyzed by flow cytometry or RNAseq [19,70]. Thus, at this regard, urine leukocytes can be potentially used as a surrogate for leukocyte infiltration in the kidney biopsies.
CD14+/CD16+ monocytes/macrophages are detected not only in the urine of LN patients, but also in other proliferative glomerulonephritis. These cells were more abundant during the acute exacerbation of renal disease, while they disappeared during remission [60]. CD14+ cells were more abundant in LN compared to non-renal SLE and healthy controls, and associated with class IV but not in class III nephritis [61]. More precisely, higher numbers of CD11c+ macrophages in the urine of patients with proliferative LN were found, with a strong association with the serum anti-dsDNA titers and chronicity indexes [62][63] (Table 1). The finding of monocytes/macrophages in urine showed a good correlation with several descriptions of infiltrates of monocytes/macrophages in kidney biopsies of LN patients [64][65][66].
M2 macrophages express CD163, an endocytic receptor of haptoglobin-hemoglobin complexes. Soluble form of the protein (sCD163) can be detected in the serum as a marker of macrophage activity [67]. Urinary sCD163 was strongly correlated with renal CD163 and histological scores [68]. Moreover, urine sCD163 increases from 6 months before the renal flare and improved the prediction of those LN patients that would achieve complete renal responses at 12 months [69]. sCD163 levels were much higher in urine than in plasma of active LN patients. After treatment, sCD163 decreased more consistently in urine than in plasma, performing better than plasma complement or anti-dsDNA titers for the differentiation between LN and non-renal SLE patients (Table 1). Urinary sCD163 concentration correlated with changes in disease activity [70]. Urine sCD163 was associated with proliferative histological classes and correlated with renal SLEDAI, but not with chronicity parameters, demonstrating again its potential as a tool to predict renal pathology [71].
CD11b is expressed in the surface of neutrophils and macrophages. Urinary levels of soluble CD11b (sCD11b) were correlated with the number of glomerular leukocytes and the histological activity (Table 1). Urinary sCD11b also decreased after glucocorticoid treatment, and performed better than sCD163 for the prediction of LN [72]. Thus, urine sCD11b can be very useful for the monitoring of LN activity and therapeutic failure, as well as for the detection of glomerular leukocyte accumulation.
Eosinophils have also been described as enriched in the urine of LN patients, compared to non-nephritis SLE. This finding was associated with higher urinary concentrations of eosinophil cationic protein (EPC) and IL-5, that could then be surrogate as markers of eosinophiluria [73].
Regarding T lymphocytes, a higher number of CD3+ cells has been reported in active LN, which correlated with SLEDAI and renal scores [74]. In a paired urine/blood cytometry study, urinary CD4+ > 800/100 mL were found exclusively in active LN patients, and their numbers normalized after treatment [75]. Urine CD8+ T cells showed a memory phenotype in active LN patients [76], and were also detectable in LN biopsies [77]. Type 1 helper (Th1) marker T-bet mRNA was found in active LN urines and tubular expression of T-bet was associated with histological activity and predicted severe flare in a longitudinal study [78]. Urinary type 17 helper (Th17) were associated with a less severe nephritis, and they were increased in blood after treatment [79]. Concerning Treg, a population of urinary CD4+CD25-Foxp3+ T cells showed a positive correlation with proteinuria in active LN patients (Table 1) [80].
Kidney-infiltrating B cells is a common finding in LN biopsies [81]. However, urine B cells have been described in LN patients in very low quantities [82], with a phenotype of Ig-secreting plasmablasts or plasma cells (PB/PC) and associated with proliferative nephritis. In addition, plasmacytoid dendritic cells (pDC) often accompanied B cells in the urine, associated with detectable urinary IFNα/β [83] (Table 1). A histological analysis revealed an infiltrate of conventional dendritic cells (cDC) and pDC with an immature phenotype in class III/IV LN patients [84], indicating a role in the immunopathogenesis of LN [64].7. Concluding Remarks
This entry is adapted from the peer-reviewed paper 10.3390/ijms22137143
References
- Lech, M.; Anders, H.J; The pathogenesis of lupus nephritis. J. Am. Soc. Nephrol 2013, 24, 1357–1366, https://doi.org/10.1681/ASN.2013010026.
- Martin Aringer; Inflammatory markers in systemic lupus erythematosus. Journal of Autoimmunity 2020, 110, 102374, 10.1016/j.jaut.2019.102374.
- Shiva Kalantari; Saeed Chashmniam; Mohsen Nafar; Zahra Zakeri; Mahmoud Parvin; Metabolomics approach reveals urine biomarkers and pathways associated with the pathogenesis of lupus nephritis.. null 2019, 22, 1288-1295, .
- Gijs Ligtenberg; Suzanne Arends; Coen A Stegeman; Karina de Leeuw; Predictors of renal flares and long-term renal outcome in patients with lupus nephritis: results from daily clinical practice.. Clin. Exp. Rheumatol 2021, , , http://www.ncbi.nlm.nih.gov/pubmed/33822705.
- Soliman, S.; Mohan, C; Lupus nephritis biomarkers. Clin. Immunol 2017, 185, 10–20., http://doi.org/10.1016/j.clim.2016.08.001.
- Anders, H.J.; Saxena, R.; Zhao, M.H.; Parodis, I.; Salmon, J.E.; Mohan, C.; Lupus nephritis. Nat. Rev. Dis. Primers 2020, 6, 7, http://doi.org/10.1038/s41572-019-0141-9.
- Amin Abedini; Yuan O. Zhu; Shatakshee Chatterjee; Gabor Halasz; Kishor Devalaraja-Narashimha; Rojesh Shrestha; Michael S. Balzer; Jihwan Park; Tong Zhou; Ziyuan Ma; et al. Urinary Single-Cell Profiling Captures the Cellular Diversity of the Kidney. Journal of the American Society of Nephrology 2021, 32, 614-627, 10.1681/asn.2020050757.
- Mok, C.C.; Mohan, C; Urinary Biomarkers in Lupus Nephritis: Are We There Yet?. Arthritis Rheumatol. 2021, 73, 194–196, http://doi.org/10.1002/art.41508.
- Feng Yu; Mark Haas; Richard Glassock; Ming-Hui Zhao; Redefining lupus nephritis: clinical implications of pathophysiologic subtypes. Nature Reviews Nephrology 2017, 13, 483-495, 10.1038/nrneph.2017.85.
- N L Meryhew; R P Messner; E M Tan; Urinary excretion of antinuclear antibodies.. The Journal of Rheumatology 1983, 10, 913–919, http://www.ncbi.nlm.nih.gov/pubmed/6363697.
- W L Picking; C Smith; R Petruci; J Scheffel; J D Levich; D A Stetler; Anti-RNA polymerase I antibodies in the urine of patients with systemic lupus erythematosus.. The Journal of Rheumatology 1990, 17, 1308–1313, .
- M Macanovic; M B Hogarth; P J Lachmann; Anti-DNA antibodies in the urine of lupus nephritis patients. Nephrology Dialysis Transplantation 1999, 14, 1418-1424, 10.1093/ndt/14.6.1418.
- Sandra A. Sciascia; Harold Dickensheets; Wendy Picking; Kristina Robson; Ng Wang; B. Hilda Ye; Liangjin Zhu; Dean A. Stetler; Autoantibodies and Autoantigens in the Urine of SLE Patients. Autoimmunity 2004, 37, 503-514, 10.1080/08916930410001713016.
- Bethany D. Harris; Srilalitha Kuruganti; Ashlesha Deshpande; Paul A. Goepfert; W. Winn Chatham; Mark R. Walter; Characterization of Type-I IFN subtype autoantibodies and activity in SLE serum and urine. Lupus 2020, 29, 1095-1105, 10.1177/0961203320935976.
- C Y Tsai; T H Wu; K H Sun; W M Lin; C L Yu; Increased excretion of soluble interleukin 2 receptors and free light chain immunoglobulins in the urine of patients with active lupus nephritis.. Annals of the Rheumatic Diseases 1992, 51, 168-172, 10.1136/ard.51.2.168.
- Masanori Hanaoka; Takahisa Gono; Yasushi Kawaguchi; Keiko Uchida; Yumi Koseki; Yasuhiro Katsumata; Hirotaka Kaneko; Kae Takagi; Hisae Ichida; Kosaku Nitta; et al. Urinary free light chain is a potential biomarker for ISN/RPS class III/IV lupus nephritis. Rheumatology 2013, 52, 2149-2157, 10.1093/rheumatology/ket108.
- John E. Hopper; Winston Sequeira; Joan Martellotto; Elaine Papagiannes; Laura Perna; John L. Skosey; Clinical relapse in systemic lupus erythematosus: Correlation with antecedent elevation of urinary free light-chain immunoglobulin. Journal of Clinical Immunology 1989, 9, 338-350, 10.1007/bf00918666.
- John E. Hopper; Joseph Golbus; Catherine Meyer; Gerrard A. Ferrer; Urine free light chains in SLE: clonal markers of B-cell activity and potential link to in vivo secreted Ig.. Journal of Clinical Immunology 2000, 20, 123-137, 10.1023/a:1006686514743.
- Mastroianni-Kirsztajn, G.; Nishida, S.K.; Pereira, A.B.; Are urinary levels of free light chains of immunoglobulins useful markers for differentiating between systemic lupus erythematosus and infection?. Nephron. Clin. Pract. 2008, 110, c258–c263, http://doi.org/10.1159/000167874.
- Carsten Paul Bramlage; Britta Froelich; Manuel Wallbach; Joan Minguet; Clemens Grupp; Cornelia Deutsch; Peter Bramlage; Michael Koziolek; Gerhard Anton Müller; The significance and predictive value of free light chains in the urine of patients with chronic inflammatory rheumatic disease. Clinical Rheumatology 2016, 35, 2939-2946, 10.1007/s10067-016-3437-0.
- Judith A. Brebner; Robert A. Stockley; Polyclonal free light chains: a biomarker of inflammatory disease or treatment target?. F1000 Medicine Reports 2013, 5, 4, 10.3410/m5-4.
- Wallace V. Epstein; Immunologic events preceding clinical exacerbation of systemic lupus erythematosus. The American Journal of Medicine 1973, 54, 631-636, 10.1016/0002-9343(73)90121-6.
- A Cooper; R Bluestone; Free immunoglobulin light chains in connective tissue diseases.. Annals of the Rheumatic Diseases 1968, 27, 537-543, 10.1136/ard.27.6.537.
- Min Chen; Mohamed R. Daha; Cees G.M. Kallenberg; The complement system in systemic autoimmune disease. Journal of Autoimmunity 2010, 34, J276-J286, 10.1016/j.jaut.2009.11.014.
- Arthur Weinstein; Roberta V. Alexander; Debra J. Zack; A Review of Complement Activation in SLE. Current Rheumatology Reports 2021, 23, 1-8, 10.1007/s11926-021-00984-1.
- Di Song; Wei-Yi Guo; Feng-Mei Wang; Yong-Zhe Li; Yan Song; Feng Yu; Ming-Hui Zhao; Complement Alternative Pathway׳s Activation in Patients With Lupus Nephritis. The American Journal of the Medical Sciences 2017, 353, 247-257, 10.1016/j.amjms.2017.01.005.
- Yoshiteru Ueda; Kohei Nagasawa; Hiroshi Tsukamoto; Takahiko Horiuchi; Seiji Yoshizawa; Tomomi Tsuru; Isao Furugo; Yoshiyuki Niho; Urinary C4 excretion in systemic lupus erythematosus. Clinica Chimica Acta 1995, 243, 11-23, 10.1016/0009-8981(95)06147-9.
- Hyunwoo Kim; Taehee Kim; Miyeon Kim; Hwa Young Lee; Yunmi Kim; Mi Seon Kang; Jinseok Kim; Activation of the alternative complement pathway predicts renal outcome in patients with lupus nephritis. Lupus 2020, 29, 862-871, 10.1177/0961203320925165.
- Anne Troldborg; Steffen Thiel; Marten Trendelenburg; Justa Friebus-Kardash; Josephine Nehring; Rudi Steffensen; Soren Hansen; Magdalena Janina Laska; Bent Deleuran; Jens Christian Jensenius; et al. The Lectin Pathway of Complement Activation in Patients with Systemic Lupus Erythematosus. The Journal of Rheumatology 2018, 45, 1136-1144, 10.3899/jrheum.171033.
- Odirlei André Monticielo; Tamara Mucenic; Ricardo Machado Xavier; João Carlos Tavares Brenol; Jose Chies; The role of mannose-binding lectin in systemic lupus erythematosus. Clinical Rheumatology 2008, 27, 413-419, 10.1007/s10067-008-0838-8.
- Birmingham, D.J.; Hebert, L.A.; The Complement System in Lupus Nephritis. Semin. Nephrol. 2015, 35, 444–454, http://doi.org/10.1016/j.semnephrol.2015.08.006.
- Susan Manzi; Joan E. Rairie; A. Betts Carpenter; Robert H. Kelly; Santhi P. Jagarlapudi; Susan M. Sereika; Thomas A. Medsger; Rosalind Ramsey-Goldman; Sensitivity and specificity of plasma and urine complement split products as indicators of lupus disease activity. Arthritis Care & Research 1996, 39, 1178-1188, 10.1002/art.1780390716.
- Yukihiro Kusunoki; Yasushi Akutsu; Noritomo Itami; Hiroyuki Tochimaru; Yasushi Nagata; Yasuo Takekoshi; Akira Sagawa; Yoshimitsu Kataoka; Shigeharu Nagasawa; Urinary Excretion of Terminal Complement Complexes in Glomerular Disease. Nephron 1991, 59, 27-32, 10.1159/000186513.
- Juan M. Mejia-Vilet; Ismael A. Gómez-Ruiz; Cristino Cruz; R. Angélica Méndez-Pérez; Roque A. Comunidad-Bonilla; Norma O. Uribe-Uribe; Carlos A. Nuñez-Alvarez; Luis E. Morales-Buenrostro; Alternative complement pathway activation in thrombotic microangiopathy associated with lupus nephritis. Clinical Rheumatology 2020, 40, 2233-2242, 10.1007/s10067-020-05499-1.
- Mariko Tamano; Yoshinobu Fuke; Morito Endo; Isao Ohsawa; Takayuki Fujita; Hiroyuki Ohi; Urinary complement factor H in renal disease.. Nephron 2002, 92, 705-707, 10.1159/000064090.
- R H Kelly; A B Carpenter; K S Sudol; S P Jagarlapudi; S Manzi; Complement C3 fragments in urine: detection in systemic lupus erythematosus patients by western blotting.. Applied and theoretical electrophoresis : the official journal of the International Electrophoresis Society 1993, 3, 265-269, .
- V S Negi; A Aggarwal; R Dayal; S Naik; R Misra; Complement degradation product C3d in urine: marker of lupus nephritis.. The Journal of Rheumatology 2000, 27, 380-383, .
- Sujata Ganguly; Sanjukta Majumder; Sandeep Kumar; Ranjan Gupta; Hafis Muhammed; Vineeta Shobha; Amita Aggarwal; Ramnath Misra; Urinary C3d is elevated in patients with active Lupus nephritis and a fall in its level after 3 months predicts response at 6 months on follow up. Lupus 2020, 29, 1800-1806, 10.1177/0961203320950019.
- Cristian C. Aragón; Raúl-Alejandro Tafúr; Ana Suárez-Avellaneda; Tatiana Martínez; Alejandra De Las Salas; Gabriel J. Tobón; Urinary biomarkers in lupus nephritis. Journal of Translational Autoimmunity 2020, 3, 100042, 10.1016/j.jtauto.2020.100042.
- Capecchi, R.; Puxeddu, I.; Pratesi, F.; Migliorini, P; New biomarkers in SLE: From bench to bedside. Rheumatology 2020, 59, v12-v18, http://doi.org/10.1093/rheumatology/keaa484.
- M Iwano; K Dohi; E Hirata; N Kurumatani; Y Horii; H Shiiki; A Fukatsu; T Matsuda; T Hirano; T Kishimoto; et al. Urinary levels of IL-6 in patients with active lupus nephritis.. Clinical Nephrology 1993, 40, 16-21, .
- Rawhya R. El-Shereef; Ahmed Lotfi; Emad A. Abdel-Naeam; Heba Tawfik; Serum and Urinary Interleukin-6 in Assessment of Renal Activity in Egyptian Patients with Systemic Lupus Erythematosus. Clinical Medicine Insights: Arthritis and Musculoskeletal Disorders 2016, 9, 29-36, 10.4137/CMAMD.S32269.
- N Bona; E Pezzarini; B Balbi; S M Daniele; M F Rossi; A L Monje; C L Basiglio; H F Pelusa; S M M Arriaga; Oxidative stress, inflammation and disease activity biomarkers in lupus nephropathy. Lupus 2020, 29, 311-323, 10.1177/0961203320904784.
- Brad H. Rovin; Xiaolan Zhang; Biomarkers for Lupus Nephritis: The Quest Continues. Clinical Journal of the American Society of Nephrology 2009, 4, 1858-1865, 10.2215/cjn.03530509.
- Lee, Y.H.; Song, G.G.; Urinary MCP-1 as a biomarker for lupus nephritis: A meta-analysis. Z. Rheumatol. 2017, 76, 357-363, http://doi.org/10.1007/s00393-016-0109-z.
- Y Ding; L-M Nie; Y Pang; W-J Wu; Ying Tan; F Yu; M-H Zhao; Composite urinary biomarkers to predict pathological tubulointerstitial lesions in lupus nephritis. Lupus 2018, 27, 1778-1789, 10.1177/0961203318788167.
- Mohamed A AboZaid; Ghada H Ahmed; Nabawia M Tawfik; Sohair K Sayed; Abeer M Ghandour; Rasha A Madkour; Serum and Urine Monocyte Chemoattractant Protein-1 as A Markers for Lupus Nephritis.. null 2020, 27, 97-107, .
- Álvaro Fernández-Ochoa; Carl Brunius; Isabel Borrás-Linares; Rosa Quirantes-Piné; María De La Luz Cádiz-Gurrea; PRECISESADS Clinical Consortium; Marta E. Alarcón Riquelme; Antonio Segura-Carretero; Metabolic Disturbances in Urinary and Plasma Samples from Seven Different Systemic Autoimmune Diseases Detected by HPLC-ESI-QTOF-MS. Journal of Proteome Research 2020, 19, 3220-3229, 10.1021/acs.jproteome.0c00179.
- Pongpratch Puapatanakul; Sonchai Chansritrakul; Paweena Susantitaphong; Thornthun Ueaphongsukkit; Somchai Eiam-Ong; Kearkiat Praditpornsilpa; Wonngarm Kittanamongkolchai; Yingyos Avihingsanon; Interferon-Inducible Protein 10 and Disease Activity in Systemic Lupus Erythematosus and Lupus Nephritis: A Systematic Review and Meta-Analysis.. International Journal of Molecular Sciences 2019, 20, 4954, 10.3390/ijms20194954.
- Juan M. Mejia‐Vilet; John P. Shapiro; Xiaolan L. Zhang; Cristino Cruz; Grant Zimmerman; R. Angélica Méndez‐Pérez; Mayra L. Cano‐Verduzco; Samir V. Parikh; Haikady N. Nagaraja; Luis E. Morales‐Buenrostro; et al. Association Between Urinary Epidermal Growth Factor and Renal Prognosis in Lupus Nephritis. Arthritis & Rheumatology 2020, 73, 244-254, 10.1002/art.41507.
- Chi Chiu Mok; Samar Soliman; Ling Yin Ho; Fatma A. Mohamed; Faten Ismail Mohamed; Chandra Mohan; Urinary angiostatin, CXCL4 and VCAM-1 as biomarkers of lupus nephritis. Arthritis Research & Therapy 2018, 20, 6, 10.1186/s13075-017-1498-3.
- Gasparin, A.A.; Pamplona Bueno de Andrade, N.; Hax, V.; Tres, G.L.; Veronese, F.V.; Monticielo, O.A; Urinary biomarkers for lupus nephritis: The role of the vascular cell adhesion molecule-1. Lupus 2019, 28, 265-272, http://doi.org/10.1177/0961203319826695.
- Andrese Aline Gasparin; Nicole Pamplona Bueno De Andrade; Vanessa Hax; Penélope Esther Palominos; Marina Siebert; Romulo Marx; Pedro Guilherme Schaefer; Francisco Veríssimo Veronese; Odirlei André Monticielo; Urinary soluble VCAM-1 is a useful biomarker of disease activity and treatment response in lupus nephritis. BMC Rheumatology 2020, 4, 67, http://doi.org/10.1186/s41927-020-00162-3.
- Ioannis Parodis; Sirisha Gokaraju; Agneta Zickert; KaMala Vanarsa; Ting Zhang; Deena Habazi; João Botto; Clara Serdoura Alves; Panagiotis Giannopoulos; Anders Larsson; et al. ALCAM and VCAM-1 as urine biomarkers of activity and long-term renal outcome in systemic lupus erythematosus. Rheumatology 2019, 59, 2237-2249, 10.1093/rheumatology/kez528.
- Huihua Ding; Cheng Lin; Jingyi Cai; Qiang Guo; Min Dai; Chandra Mohan; Nan Shen; Urinary activated leukocyte cell adhesion molecule as a novel biomarker of lupus nephritis histology. Arthritis Research & Therapy 2020, 22, 1-9, 10.1186/s13075-020-02209-9.
- Run-Nan Guo Liu; Qian-Yao Cheng; Hao-Yue Zhou; Bao-Zhu Li; Dong-Qing Ye; Elevated Blood and Urinary ICAM-1 is a Biomarker for Systemic Lupus Erythematosus: A Systematic Review and Meta-Analysis. Immunological Investigations 2019, 49, 15-31, 10.1080/08820139.2019.1624769.
- Li Liu; Ran Wang; Huihua Ding; Lei Tian; Ting Gao; Chunde Bao; The utility of urinary biomarker panel in predicting renal pathology and treatment response in Chinese lupus nephritis patients. PLOS ONE 2020, 15, e0240942, 10.1371/journal.pone.0240942.
- Prasad Devarajan; Review: Neutrophil gelatinase-associated lipocalin: A troponin-like biomarker for human acute kidney injury. Nephrology 2010, 15, 419-428, 10.1111/j.1440-1797.2010.01317.x.
- Mohamed S. El Shahawy; Mahmoud H. Hemida; Hafez A. Abdel-Hafez; Tarek Z. El-Baz; Abdel-Wahab M. Lotfy; Tarek M. Emran; Urinary neutrophil gelatinase-associated lipocalin as a marker for disease activity in lupus nephritis. Scandinavian Journal of Clinical and Laboratory Investigation 2018, 78, 264-268, 10.1080/00365513.2018.1449242.
- Osamu Hotta; Naoko Yusa; Michio Ooyama; Kazuko Unno; Takashi Furuta; Yoshio Taguma; Detection of urinary macrophages expressing the CD16 (FcγRIII) molecule: A novel marker of acute inflammatory glomerular injury. Kidney International 1999, 55, 1927-1934, 10.1046/j.1523-1755.1999.00431.x.
- Abeer Ali Abdelati; Nouran Y. Eshak; Hanaa M. Donia; Amira H. El-Girby; Urinary Cellular Profile as a Biomarker for Lupus Nephritis. JCR: Journal of Clinical Rheumatology 2020, Publish Ah, -, 10.1097/rhu.0000000000001553.
- Jihye Kim; Jung Sun Lee; Heounjeong Go; Joon Seo Lim; Ji Seon Oh; Yong-Gil Kim; Chang-Keun Lee; Bin Yoo; Seokchan Hong; Clinical and histological significance of urinary CD11c+ macrophages in lupus nephritis. Arthritis Research & Therapy 2020, 22, 1-8, 10.1186/s13075-020-02265-1.
- Jihye Kim; Ji Hye Jeong; Jaehyung Jung; Hanwool Jeon; Seungjoo Lee; Joon Seo Lim; Heounjeong Go; Ji Seon Oh; Yong-Gil Kim; Chang-Keun Lee; et al. Immunological characteristics and possible pathogenic role of urinary CD11c+ macrophages in lupus nephritis. Rheumatology 2020, 59, 2135-2145, 10.1093/rheumatology/keaa053.
- Naomi I. Maria; Anne Davidson; Renal Macrophages and Dendritic Cells in SLE Nephritis. Current Rheumatology Reports 2017, 19, 81, 10.1007/s11926-017-0708-y.
- Yiling Cao; Weihao Tang; Wanxin Tang; Immune cell infiltration characteristics and related core genes in lupus nephritis: results from bioinformatic analysis. BMC Immunology 2019, 20, 1-12, 10.1186/s12865-019-0316-x.
- Jeeba Kuriakose; Vanessa Redecke; Cliff Guy; Jingran Zhou; Ruiqiong Wu; Sirish K. Ippagunta; Heather Tillman; Patrick D. Walker; Peter Vogel; Hans Häcker; et al. Patrolling monocytes promote the pathogenesis of early lupus-like glomerulonephritis. Journal of Clinical Investigation 2019, 129, 2251-2265, 10.1172/jci125116.
- Holger Jon Møller; Marianne Jensby Nielsen; Maciej Bogdan Maniecki; Mette Madsen; Søren Kragh Moestrup; Soluble macrophage-derived CD163: A homogenous ectodomain protein with a dissociable haptoglobin–hemoglobin binding. Immunobiology 2010, 215, 406-412, 10.1016/j.imbio.2009.05.003.
- Nobuhide Endo; Naotake Tsuboi; Kazuhiro Furuhashi; Yiqin Shi; Qiuna Du; Tomoko Abe; Mayuko Hori; Takahiro Imaizumi; Hangsoo Kim; Takayuki Katsuno; et al. Urinary soluble CD163 level reflects glomerular inflammation in human lupus nephritis. Nephrology Dialysis Transplantation 2016, 31, 2023-2033, 10.1093/ndt/gfw214.
- Juan M. Mejia-Vilet; Xiaolan L. Zhang; Cristino Cruz; Mayra L. Cano-Verduzco; John P. Shapiro; Haikady N. Nagaraja; Luis E. Morales-Buenrostro; Brad H. Rovin; Urinary Soluble CD163: a Novel Noninvasive Biomarker of Activity for Lupus Nephritis. Journal of the American Society of Nephrology 2020, 31, 1335-1347, 10.1681/asn.2019121285.
- Ranjan Gupta; Akhilesh Yadav; Amita Aggarwal; Urinary soluble CD163 is a good biomarker for renal disease activity in lupus nephritis. Clinical Rheumatology 2020, 40, 941-948, 10.1007/s10067-020-05343-6.
- Ting Zhang; Hao Li; KaMala Vanarsa; Gabriel Gidley; Chi Chiu Mok; Michelle Petri; Ramesh Saxena; Chandra Mohan; Association of Urine sCD163 With Proliferative Lupus Nephritis, Fibrinoid Necrosis, Cellular Crescents and Intrarenal M2 Macrophages. Frontiers in Immunology 2020, 11, 671, 10.3389/fimmu.2020.00671.
- Akimitsu Kitagawa; Naotake Tsuboi; Yuki Yokoe; Takayuki Katsuno; Hidekazu Ikeuchi; Hiroshi Kajiyama; Nobuhide Endo; Yuriko Sawa; Junya Suwa; Yutaka Sugiyama; et al. Urinary levels of the leukocyte surface molecule CD11b associate with glomerular inflammation in lupus nephritis. Kidney International 2019, 95, 680-692, 10.1016/j.kint.2018.10.025.
- Tereza Neuma Souza Brito; Maria José Vilar; José Bruno Almeida; Ana Luiza Souza Brito Faria; Sarah Dantas Viana Medeiros; Maria Carmo Cardoso Medeiros; Edna Marques Araújo Silva; Vanessa Marques Araújo Silva; Luanda Bárbara F Canário Souza; Luisa Karla P Arruda; et al. Measuring eosinophiluria, urinary eosinophil cationic protein and urinary interleukin-5 in patients with Lupus Nephritis.. Allergy, Asthma & Clinical Immunology 2014, 10, 61, 10.1186/s13223-014-0061-x.
- R W Y Chan; F M M Lai; E K M Li; L S Tam; K Y Chung; K M Chow; P K T Li; C C Szeto; Urinary mononuclear cell and disease activity of systemic lupus erythematosus. Lupus 2006, 15, 262-267, 10.1191/0961203306lu2287oa.
- Philipp Enghard; Claudia Rieder; Katharina Kopetschke; J. R. Klocke; Reinmar Undeutsch; Robert Biesen; Duska Dragun; Maik Gollasch; Udo Schneider; Karlfried Aupperle; et al. Urinary CD4 T cells identify SLE patients with proliferative lupus nephritis and can be used to monitor treatment response. Annals of the Rheumatic Diseases 2013, 73, 277-283, 10.1136/annrheumdis-2012-202784.
- Sebastian Dolff; Wayel H Abdulahad; Suzanne Arends; Marcory Crf Van Dijk; Pieter C Limburg; Cees Gm Kallenberg; Marc Bijl; Urinary CD8+ T-cell counts discriminate between active and inactive lupus nephritis. Arthritis Research & Therapy 2013, 15, R36-R36, 10.1186/ar4189.
- Sebastian Dolff; Wayel H. Abdulahad; Marcory C. R. F. Van Dijk; Pieter C. Limburg; Cees G. M. Kallenberg; Marc Bijl; Urinary T cells in active lupus nephritis show an effector memory phenotype. Annals of the Rheumatic Diseases 2010, 69, 2034-2041, 10.1136/ard.2009.124636.
- R. W.-Y. Chan; F. M.-M. Lai; E. K.-M. Li; Lai-Shan Tam; K.-M. Chow; P. K.-T. Li; C.-C. Szeto; Expression of T-bet, a type 1 T-helper cell transcription factor, in the urinary sediment of lupus patients predicts disease flare. Rheumatology 2007, 46, 44-48, 10.1093/rheumatology/kel192.
- Mesquita, D., Jr.; Kirsztajn, G.M.; Franco, M.F.; Reis, L.A.; Perazzio, S.F.; Mesquita, F.V.; Ferreira, V.D.S.; Andrade, L.E.C.; de Souza, A.W.S.; CD4(+) T helper cells and regulatory T cells in active lupus nephritis: An imbalance towards a predominant Th1 response?. Clin. Exp. Immunol. 2018, 191, 50-59, http://doi.org/10.1111/cei.13050.
- Bonelli, M.; Goschl, L.; Bluml, S.; Karonitsch, T.; Steiner, C.W.; Steiner, G.; Smolen, J.S.; Scheinecker, C.; CD4(+)CD25(-)Foxp3(+) T cells: A marker for lupus nephritis?. Arthritis Res. Ther. 2014, 16, R104, http://doi.org/10.1186/ar4553.
- Arnon Arazi; Deepak A. Rao; Celine C. Berthier; Anne Davidson; Yanyan Liu; Paul J. Hoover; Adam Chicoine; Thomas M. Eisenhaure; A. Helena Jonsson; Shuqiang Li; et al. The immune cell landscape in kidneys of lupus nephritis patients. Nat. Immunol. 2019, 20, 902-914, 10.1101/363051.
- Martina Bertolo; Sabine Baumgart; Pawel Durek; Anette Peddinghaus; Henrik Mei; Thomas Rose; Philipp Enghard; Andreas Grützkau; Deep Phenotyping of Urinary Leukocytes by Mass Cytometry Reveals a Leukocyte Signature for Early and Non-Invasive Prediction of Response to Treatment in Active Lupus Nephritis. Frontiers in Immunology 2020, 11, 256, 10.3389/fimmu.2020.00256.
- Eric Scott; Mary Anne Dooley; Barbara J. Vilen; Stephen H. Clarke; Immune cells and type 1 IFN in urine of SLE patients correlate with immunopathology in the kidney. Clinical Immunology 2016, 168, 16-24, 10.1016/j.clim.2016.04.005.
- Nicoletta Fiore; Giuseppe Castellano; Antonella Blasi; Carmen Capobianco; Antonia Loverre; Vincenzo Montinaro; Giuseppe Stefano Netti; Diletta Torres; Carlo Manno; Giuseppe Grandaliano; et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Molecular Immunology 2008, 45, 259-265, 10.1016/j.molimm.2007.04.029.