The inhibitors of β-lactamases (BLI) have gained a prominent role in the safeguard of beta-lactams. In the last years, new β-lactam–BLI combinations have been registered or are still under clinical evaluation, demonstrating their effectiveness to treat complicated infections. It is also noteworthy that the pharmacokinetics of BLIs partly matches that of β-lactams companions, meaning that some clinical situations, as well as renal impairment and renal replacement therapies, may alter the disposition of both drugs. Common pharmacokinetic characteristics, linear pharmacokinetics across a wide range of doses, and known pharmacokinetic/pharmacodynamic parameters may guide modifications of dosing regimens for both β-lactams and BLIs. However, comorbidities (i.e., burns, diabetes, cancer) and severe changes in individual pathological conditions (i.e., acute renal impairment, sepsis) could make dose adaptation difficult, because the impact of those factors on BLI pharmacokinetics is partly known. Therapeutic drug monitoring protocols may overcome those issues and offer strategies to personalize drug doses in the intensive care setting. Further prospective clinical trials are warranted to improve the use of BLIs and their β-lactam companions in severe and complicated infections.
- avibactam
- vaborbactam
- relebactam
- durlobactam
- pharmacokinetics
- pharmacokinetics/pharmacodynamics
1. Introduction
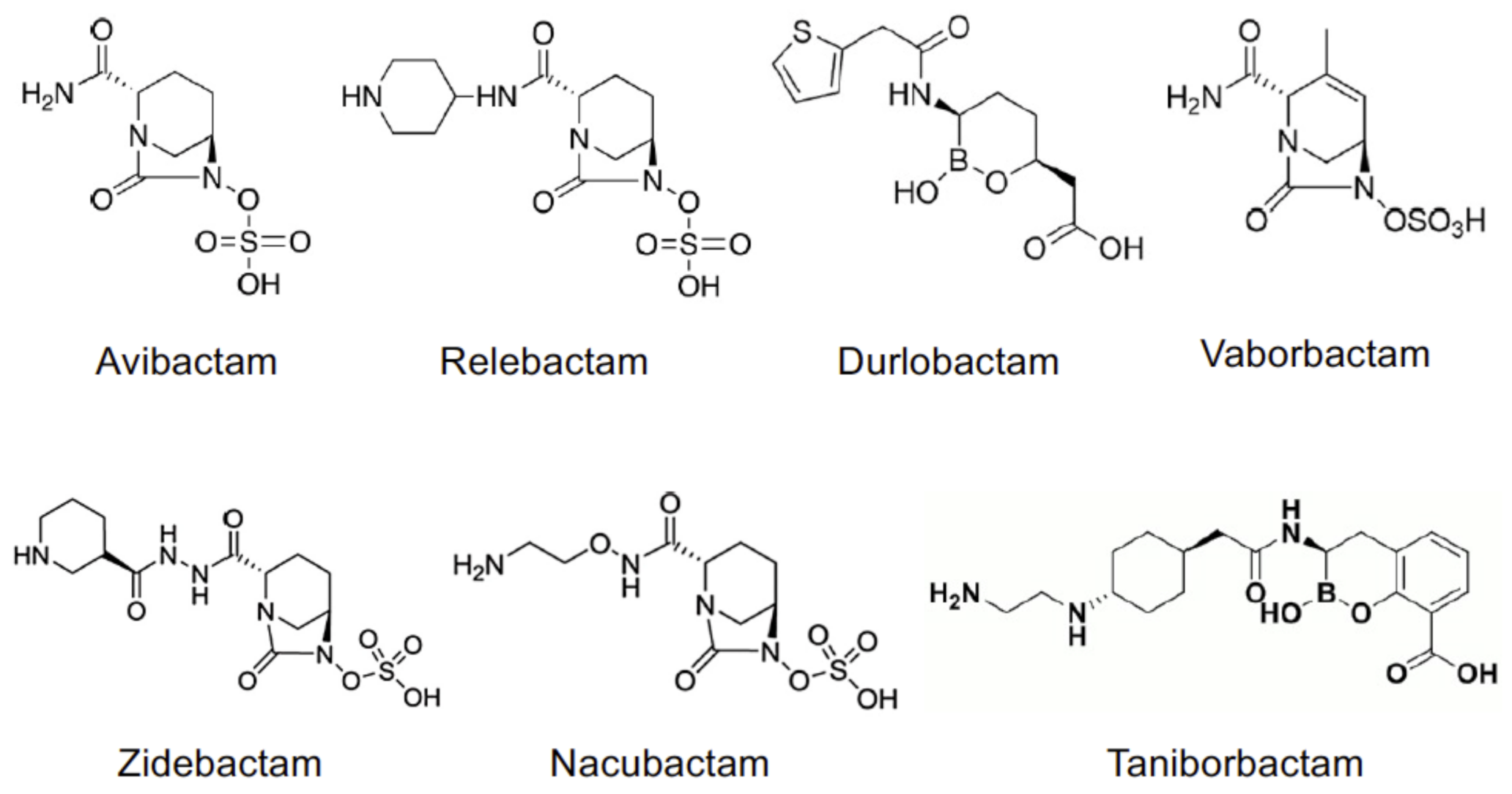 Figure 1. Chemical structures of non-beta-lactam BLIs.
Figure 1. Chemical structures of non-beta-lactam BLIs.2. Pharmacokinetics of BLIs
| Drug | CL 1 (L/h) | Vd (L) | t1/2 (h) | PPB (%) | References |
|---|---|---|---|---|---|
| AVI | 1.59 | 18.0 | 2.0 | 8 | [11] |
| CAZ | 1.54 | 22.0 | 2.0 | 10 | [11] |
| AZT | 140 | 29.4 | 1.7 | 77 | [12][13] |
| CEF | 1.8–3.0 | 28.3 | 2.5 | 20 | [14] |
| VAB | 10.5 | 19.0 | 2.25 | 33 | [15][16][17] |
| MER | 7.7 | 21.0 | 2.30 | 2 | [15][16][17] |
| REL | 8.1 | 21 | 1.7 | 22 | [15][18] |
| IMI | 8.4 | 21.7 | 1.1 | 20 | [19][18][20] |
| DUR | 10.3 | 31.6 | 2.5 | - | [20] |
| SUL | 2.4 | 12.0 | 1.8 | 38 | [20][21] |
| ZID | 7.4 | 17.4 | 1.9 | <15 | [22] |
| NAC | 8.8 | 20.6 | 2.4 | - | [23] |
| TAN | 5.8 | 30–50 | 6.5 | - | [24][25] |
| Drugs and Dosage | Clinical Use | Therapeutic Indications (Duration of Treatment) |
Pediatric Use | Ref. |
|---|---|---|---|---|
| CAZ/AVI 1 2/0.5 g q8h 2-h IV infusion |
YES | cIAI (5–14 days) cUTI (5–10 days) Pyelonephritis (5–10 days) HAP (7–14 days) VAP (7–14 days) Aerobic G- infections (variable) |
YES (age ≥3 months) |
[11] |
| MER/VAB 2/2 g q8h 3-h IV infusion |
YES | cIAI (5–10 days) cUTI (5–10 days) Pyelonephritis (5–10 days) HAP (7–14 days) VAP (7–14 days) Aerobic G- infections (variable) Bacteremia (variable) |
NO | [17] |
| REL/IMI/CIL 0.25/0.5/0.5 g q6h 0.5-h IV infusion |
YES | HAP (7–14 days) VAP (7–14 days) Infections from Aerobic G- (variable) Bacteremia (variable) |
NO | [18] |
| REL/IMI 0.25/0.5 g q6h 0.5-h IV infusion |
NO | HAP (variable) VAP (variable) cUTI (variable) cIAI (variable) |
- | [37][38][39] |
| DUR/SUL | NO | Phase III studies: cUTI, HAP, VAP | - | [40][41] |
| DUR/IMI/CIL | NO | Studies in HV | - | [28] |
| ZID/CEF | NO | Phase III studies: cUTI, HAP, VAP | - | [41] |
| NAC/CEF or AZT | NO | Phase I studies: cUTI | - | [41] |
| TAN/CEF | NO | Phase III studies: cUTI | - | [41] |
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics10070769
References
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62.
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500.
- Livermore, D.M. Determinants of the Activity of Beta-Lactamase Inhibitor Combinations. J. Antimicrob. Chemother. 1993, 31 (Suppl. A), 9–21.
- Berkhout, J.; Melchers, M.J.; van Mil, A.C.; Seyedmousavi, S.; Lagarde, C.M.; Schuck, V.J.; Nichols, W.W.; Mouton, J.W. Pharmacodynamics of Ceftazidime and Avibactam in Neutropenic Mice with Thigh or Lung Infection. Antimicrob. Agents Chemother. 2016, 60, 368–375.
- Abdul-Aziz, M.H.; Lipman, J.; Akova, M.; Bassetti, M.; De Waele, J.J.; Dimopoulos, G.; Dulhunty, J.; Kaukonen, K.-M.; Koulenti, D.; Martin, C.; et al. Is Prolonged Infusion of Piperacillin/Tazobactam and Meropenem in Critically Ill Patients Associated with Improved Pharmacokinetic/Pharmacodynamic and Patient Outcomes? An Observation from the Defining Antibiotic Levels in Intensive Care Unit Patients (DA. J. Antimicrob. Chemother. 2016, 71, 196–207.
- Vardakas, K.Z.; Voulgaris, G.L.; Maliaros, A.; Samonis, G.; Falagas, M.E. Prolonged versus Short-Term Intravenous Infusion of Antipseudomonal β-Lactams for Patients with Sepsis: A Systematic Review and Meta-Analysis of Randomised Trials. Lancet Infect. Dis. 2018, 18, 108–120.
- Lister, P.D.; Prevan, A.M.; Sanders, C.C. Importance of Beta-Lactamase Inhibitor Pharmacokinetics in the Pharmacodynamics of Inhibitor-Drug Combinations: Studies with Piperacillin-Tazobactam and Piperacillin-Sulbactam. Antimicrob. Agents Chemother. 1997, 41, 721–727.
- Vanscoy, B.; Mendes, R.E.; McCauley, J.; Bhavnani, S.M.; Bulik, C.C.; Okusanya, O.O.; Forrest, A.; Jones, R.N.; Friedrich, L.V.; Steenbergen, J.N.; et al. Pharmacological Basis of β-Lactamase Inhibitor Therapeutics: Tazobactam in Combination with Ceftolozane. Antimicrob. Agents Chemother. 2013, 57, 5924–5930.
- Veiga, R.P.; Paiva, J.-A. Pharmacokinetics-Pharmacodynamics Issues Relevant for the Clinical Use of Beta-Lactam Antibiotics in Critically Ill Patients. Crit. Care 2018, 22, 233.
- Falcone, M.; Daikos, G.L.; Tiseo, G.; Bassoulis, D.; Giordano, C.; Galfo, V.; Leonildi, A.; Tagliaferri, E.; Barnini, S.; Sani, S.; et al. Efficacy of Ceftazidime-Avibactam plus Aztreonam in Patients with Bloodstream Infections Caused by MBL- Producing Enterobacterales. Clin. Infect. Dis. 2020.
- European Medicines Agency (EMA). Summary of Product Characteristics—Ceftazidime/Avibactam—535. Available online: (accessed on 13 June 2021).
- European Medicines Agency (EMA). Summary of Product Characteristics—Aztreonam—537. Available online: (accessed on 13 June 2021).
- Mattie, H. Clinical Pharmacokinetics of Aztreonam. Clin. Pharmacokinet. 1994, 26, 99–106.
- European Medicines Agency (EMA). Summary of Product Characteristics—Ceftarolin—541. Available online: (accessed on 13 June 2021).
- Zhanel, G.G.; Lawrence, C.K.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Zhanel, M.; Lagacé-Wiens, P.R.S.; Walkty, A.; Denisuik, A.; Golden, A.; et al. Imipenem-Relebactam and Meropenem-Vaborbactam: Two Novel Carbapenem-β-Lactamase Inhibitor Combinations. Drugs 2018, 78, 65–98.
- Patel, T.S.; Pogue, J.M.; Mills, J.P.; Kaye, K.S. Meropenem–Vaborbactam: A New Weapon in the War against Infections Due to Resistant Gram-Negative Bacteria. Future Microbiol. 2018, 13, 971–983.
- European Medicines Agency (EMA). Summary of Product Characteristics—Vaborbactam/Meropenem—545. Available online: (accessed on 13 June 2021).
- European Medicines Agency (EMA). Summary of Product Characteristics—Relebactam/Imipenem/Cilastatin—547. Available online: (accessed on 13 June 2021).
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692.
- Sagan, O.; Yakubsevitch, R.; Yanev, K.; Fomkin, R.; Stone, E.; Hines, D.; O’Donnell, J.; Miller, A.; Isaacs, R.; Srinivasan, S. Pharmacokinetics and Tolerability of Intravenous Sulbactam-Durlobactam with Imipenem-Cilastatin in Hospitalized Adults with Complicated Urinary Tract Infections, Including Acute Pyelonephritis. Antimicrob. Agents Chemother. 2020, 64.
- Foulds, G.; Stankewich, J.P.; Marshall, D.C.; O’Brien, M.M.; Hayes, S.L.; Weidler, D.J.; McMahon, F.G. Pharmacokinetics of Sulbactam in Humans. Antimicrob. Agents Chemother. 1983, 23, 692–699.
- Rodvold, K.A.; Gotfried, M.H.; Chugh, R.; Gupta, M.; Patel, A.; Chavan, R.; Yeole, R.; Friedland, H.D.; Bhatia, A. Plasma and Intrapulmonary Concentrations of Cefepime and Zidebactam Following Intravenous Administration of WCK 5222 to Healthy Adult Subjects. Antimicrob. Agents Chemother. 2018, 62.
- Mallalieu, N.L.; Winter, E.; Fettner, S.; Patel, K.; Zwanziger, E.; Attley, G.; Rodriguez, I.; Kano, A.; Salama, S.M.; Bentley, D.; et al. Safety and Pharmacokinetic Characterization of Nacubactam, a Novel β-Lactamase Inhibitor, Alone and in Combination with Meropenem, in Healthy Volunteers. Antimicrob. Agents Chemother. 2020, 64.
- Geibel, B.; Dowell, J.A.; Marbury, T.C.; Smith, W.; McGovern, P.C.; Richards, C.; Henkel, T. Pharmacokinetics and Safety of Cefepime-Taniborbactam (Formerly Cefepime/VNRX-5133) in Subjects with Renal Impairment. Open Forum Infect. Dis. 2020, 7, S670.
- Geibel, B.; Dowell, J.; Dickerson, D.; Henkel, T. A Randomized, Double-Blind, Placebo-Controlled Study of the Safety and Pharmacokinetics of Single and Repeat Doses of VNRX-5133 in Healthy Subjects. Open Forum Infect. Dis. 2018, 5, S431.
- Ho, S.; Nguyen, L.; Trinh, T.; MacDougall, C. Recognizing and Overcoming Resistance to New Beta-Lactam/Beta-Lactamase Inhibitor Combinations. Curr. Infect. Dis. Rep. 2019, 21, 39.
- Nichols, W.W.; Newell, P.; Critchley, I.A.; Riccobene, T.; Das, S. Avibactam Pharmacokinetic/Pharmacodynamic Targets. Antimicrob. Agents Chemother. 2018, 62.
- Rhee, E.G.; Rizk, M.L.; Calder, N.; Nefliu, M.; Warrington, S.J.; Schwartz, M.S.; Mangin, E.; Boundy, K.; Bhagunde, P.; Colon-Gonzalez, F.; et al. Pharmacokinetics, Safety, and Tolerability of Single and Multiple Doses of Relebactam, a β-Lactamase Inhibitor, in Combination with Imipenem and Cilastatin in Healthy Participants. Antimicrob. Agents Chemother. 2018, 62.
- Falcone, M.; Menichetti, F.; Cattaneo, D.; Tiseo, G.; Baldelli, S.; Galfo, V.; Leonildi, A.; Tagliaferri, E.; Di Paolo, A.; Pai, M.P. Pragmatic Options for Dose Optimization of Ceftazidime/Avibactam with Aztreonam in Complex Patients. J. Antimicrob. Chemother. 2021, 76, 1025–1031.
- Vena, A.; Giacobbe, D.R.; Castaldo, N.; Cattelan, A.; Mussini, C.; Luzzati, R.; De Rosa, F.G.; Del Puente, F.; Mastroianni, C.M.; Cascio, A.; et al. Clinical Experience with Ceftazidime-Avibactam for the Treatment of Infections Due to Multidrug-Resistant Gram-Negative Bacteria Other than Carbapenem-Resistant Enterobacterales. Antibiotics 2020, 9, 71.
- Rubino, C.M.; Bhavnani, S.M.; Loutit, J.S.; Lohse, B.; Dudley, M.N.; Griffith, D.C. Single-Dose Pharmacokinetics and Safety of Meropenem-Vaborbactam in Subjects with Chronic Renal Impairment. Antimicrob. Agents Chemother. 2018, 62.
- O’Donnell, J.; Preston, R.A.; Mamikonyan, G.; Stone, E.; Isaacs, R. Pharmacokinetics, Safety, and Tolerability of Intravenous Durlobactam and Sulbactam in Subjects with Renal Impairment and Healthy Matched Control Subjects. Antimicrob. Agents Chemother. 2019, 63.
- Bhagunde, P.; Patel, P.; Lala, M.; Watson, K.; Copalu, W.; Xu, M.; Kulkarni, P.; Young, K.; Rizk, M.L. Population Pharmacokinetic Analysis for Imipenem-Relebactam in Healthy Volunteers and Patients with Bacterial Infections. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 748–758.
- Soukup, P.; Faust, A.C.; Edpuganti, V.; Putnam, W.C.; McKinnell, J.A. Steady-State Ceftazidime-Avibactam Serum Concentrations and Dosing Recommendations in a Critically Ill Patient Being Treated for Pseudomonas Aeruginosa Pneumonia and Undergoing Continuous Venovenous Hemodiafiltration. Pharmacotherapy 2019, 39, 1216–1222.
- Li, J.; Lovern, M.; Green, M.L.; Chiu, J.; Zhou, D.; Comisar, C.; Xiong, Y.; Hing, J.; MacPherson, M.; Wright, J.G.; et al. Ceftazidime-Avibactam Population Pharmacokinetic Modeling and Pharmacodynamic Target Attainment Across Adult Indications and Patient Subgroups. Clin. Transl. Sci. 2019, 12, 151–163.
- Boidin, C.; Moshiri, P.; Dahyot-Fizelier, C.; Goutelle, S.; Lefeuvre, S. Pharmacokinetic Variability of Beta-Lactams in Critically Ill Patients: A Narrative Review. Anaesth. Crit. Care Pain Med. 2020, 39, 87–109.
- Motsch, J.; Murta de Oliveira, C.; Stus, V.; Köksal, I.; Lyulko, O.; Boucher, H.W.; Kaye, K.S.; File, T.M.; Brown, M.L.; Khan, I.; et al. RESTORE-IMI 1: A Multicenter, Randomized, Double-Blind Trial Comparing Efficacy and Safety of Imipenem/Relebactam vs Colistin Plus Imipenem in Patients With Imipenem-Nonsusceptible Bacterial Infections. Clin. Infect. Dis. 2020, 70, 1799–1808.
- Brown, M.L.; Motsch, J.; Kaye, K.S.; File, T.M.; Boucher, H.W.; Vendetti, N.; Aggrey, A.; Joeng, H.-K.; Tipping, R.W.; Du, J.; et al. Evaluation of Renal Safety Between Imipenem/Relebactam and Colistin Plus Imipenem in Patients with Imipenem-Nonsusceptible Bacterial Infections in the Randomized, Phase 3 RESTORE-IMI 1 Study. Open Forum Infect. Dis. 2020, 7.
- Kaye, K.S.; Boucher, H.W.; Brown, M.L.; Aggrey, A.; Khan, I.; Joeng, H.-K.; Tipping, R.W.; Du, J.; Young, K.; Butterton, J.R.; et al. Comparison of Treatment Outcomes between Analysis Populations in the RESTORE-IMI 1 Phase 3 Trial of Imipenem-Cilastatin-Relebactam versus Colistin plus Imipenem-Cilastatin in Patients with Imipenem-Nonsusceptible Bacterial Infections. Antimicrob. Agents Chemother. 2020, 64.
- McLeod, S.M.; Moussa, S.H.; Hackel, M.A.; Miller, A.A. In Vitro Activity of Sulbactam-Durlobactam against Acinetobacter Baumannii—Calcoaceticus Complex Isolates Collected Globally in 2016 and 2017. Antimicrob. Agents Chemother. 2020, 64.
- PEW Trusts. Available online: (accessed on 13 June 2021).
- Abdul-Aziz, M.H.; Alffenaar, J.-W.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.-A.; Pea, F.; Sjovall, F.; et al. Antimicrobial Therapeutic Drug Monitoring in Critically Ill Adult Patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153.
- Sillén, H.; Mitchell, R.; Sleigh, R.; Mainwaring, G.; Catton, K.; Houghton, R.; Glendining, K. Determination of Avibactam and Ceftazidime in Human Plasma Samples by LC-MS. Bioanalysis 2015, 7, 1423–1434.
- Abdulla, A.; Bahmany, S.; Wijma, R.A.; van der Nagel, B.C.H.; Koch, B.C.P. Simultaneous Determination of Nine β-Lactam Antibiotics in Human Plasma by an Ultrafast Hydrophilic-Interaction Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1060, 138–143.
- Sutherland, C.A.; Nicolau, D.P. Development of an HPLC Method for the Determination of Meropenem/Vaborbactam in Biological and Aqueous Matrixes. J. Chromatogr. Sci. 2020, 58, 726–730.
- Sarli, V.; Ciofi, L.; Lastella, M.; Muscatello, B.; Pisaturo, F.; Paolilli, O.; Luci, G.; Cucchiara, F.; Pellegrini, G.; Bocci, G.; et al. Appropriateness of Repetitive Therapeutic Drug Monitoring and Laboratory Turnaround Time. Clin. Chem. Lab. Med. 2019, 57, e331–e333.
- Sprague, D.A.; Ensom, M.H.H. Limited-Sampling Strategies for Anti-Infective Agents: Systematic Review. Can. J. Hosp. Pharm. 2009, 62, 392–401.
- Di Paolo, A.; Tascini, C.; Polillo, M.; Gemignani, G.; Nielsen, E.I.; Bocci, G.; Karlsson, M.O.; Menichetti, F.; Danesi, R. Population Pharmacokinetics of Daptomycin in Patients Affected by Severe Gram-Positive Infections. Int. J. Antimicrob. Agents 2013, 42, 250–255.