Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Hemorphins are endogenous peptides that are also known as “non-classical” or “atypical” opioid peptides. They are produced under physiologicalor pathological(inflammation) states by hemoglobin proteolysis. They can be released from almost any of the hemoglobin chains (beta-, kappa-, delta-, or epsilon-chain) except the alpha chain.
- hemoglobin
- hemorphins
- analysis
- proteolytic enzymes
- sequencing
- mass spectrometry
- identification
- pain
- alcohol
- opioids
- receptors
1. Introduction
Biochemical analysis has confirmed the presence of a substantial concentration of hemorphins in the human pituitary gland [1], bovine hypothalamus [2], bovine brain [3], adrenal glands [4], as well as other organs [5] and body fluids [6]. The high stability of these peptides in plasma or tissues and their wide distribution may suggest significant role of these peptides in various processes [7].
The shortest sequence of hemorphins maintaining its binding to opioid receptors is Tyr-Pro-Trp-Thr. First report, published by the A. Herz′s group, described identification of hemorphin-4 (H-4) and H-5 as they were released from bovine blood by gastrointestinal enzymes [8]. This work was performed nearly accidentally, during identification of closely eluted cytochrophin-4 and, at that time, only amino acid analysis and Edman degradation were available, but these techniques were sufficient for a complete identification of the in vitro released products.
Hemorphins can be produced in vitro by endogenous lysosomal proteases [9], pepsin [10], pancreatic elastase [11] or cathepsin D [12][13].
It is still uncertain which enzymes are responsible for the generation of hemorphins from hemoglobin chains. It has been postulated that these peptides may also be released from other, hitherto unknown, proteins [7]. A recent, thorough search for the sequence of LVV-H-7 by the UniProtKB 2020_06 knowledgebase shows unequivocally that hemorphins are, indeed, derived from hemoglobin chains.
Opioid properties of several peptides, including hemorphins, beta-casomorphins and cytochrophins, were described by Zadina et al. [14] as showing their ability to inhibit binding of the brain peptide Tyr-MIF-1 (Tyr-Pro-Leu-Gly-NH2) to its high affinity sites in rat brain. In general, all hemorphin fragments bind to the mu-opioid receptor. However, various sequences may also bind to other types of receptors, such as delta or sigma sites, though to a lower extent. Further studies have indicated that these peptides may also maintain a balance between opiate and antiopiate activities. The peptides mentioned above were artificially generated from hemoglobin. The first report on the naturally occurring peptide was presented by Glamsta et al. [6].
Particular focus has been aimed at the identification of endogenous LVV-H7 in body fluids. Under physiological conditions, LVV-H7 is not detectable in human cerebrospinal fluid (hCSF). In contrast, cerebral hemorrhage triggers the release of this peptide to a very high level in CSF (estimated at 115–300 pmol/mL). This observation has led to the isolation and identification of this sequence by the gas-phase sequencing and also by direct sequencing by mass spectrometry [6], and also without extensive preseparation [15].
Here, we must also underline the multiple role of hemoglobin in the body, which arises from such studies. Major functions of the protein are oxygen transport and removal of carbon dioxide. Other roles of hemoglobin, discovered much later, are based on the release of opioid peptides—hemorphins and longer sequences—hemocidins, possessing antibacterial properties [16].
2. Pharmacology of Hemorphins
Various pharmacological effects of hemorphins are related to their high affinity to μ -, δ – and κ – opioid receptors [7][1], angiotensin (Ang) IV (AT4) receptor, bombesin subtype 3 receptor (hBRS-3) [17] or corticotropin releasing factor (CRF) receptors [18]. However, the binding of hemorphins to opioid receptors is much lower than that to classical opioid peptides. Simultaneously, some studies suggest that these peptides may affect the opioid receptor system in a manner similar to classic opiates, or endogenous opioid peptides (enkephalins and endorphins), due to the relatively high content of these hemoglobin fragments in the tissues [3][19].
Numerous functional interactions between hemorphins and β-endorphin, growth hormone, prolactin [7], substance P (SP) [20], neuropeptide Y, Met-ENK-Arg-Phe [21] or CRF [18][22] have been observed. It should be emphasized that hemorphins, as members of the endogenous protective system of the organism, play a significant role mainly in response to pathophysiological conditions (e.g., stress, inflammation, cancer). In this case, hemorphins, similar to other pleiotropic neuropeptides [23], serve as one of the homeostatic factors that activate compensatory systems within the organism. This effect could be observed as the occurrence of interactions between CRP, opioids and hemorphins in the hypothalamus [2] and pituitary gland [1] in response to stressful conditions [7].
3. Hemorphin Analogues
The series of hemorphin analogues have been synthesized and the structure–activity relationship of these compounds has been elucidated by Todorov’s group [24]. The synthesis of peptides was performed manually using 9-fluorenylmethoxycarbonyl (Fmoc) solid phase synthesis. The basic concept of the method is the stepwise construction of a peptide chain assembled on an insoluble polymeric support. Chain elongation starts at the C-terminal residue. The peptide formation goes through a repetitive amidation reaction between an activated carboxylic group of one amino acid and the amino group of the second one. Here, the coupling was accomplished via activation with 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) and hydroxybenzotriazole (HOBt) in the presence of N,N-Diisopropylethylamine (DIEA). Each incoming amino acid was protected at the α-amino moiety by the Fmoc group to prevent formation of a peptide bond at that site. After coupling, the Fmoc group was removed using a 20% piperidine solution and the process was repeated. Due to the possibility of unintended reactions during synthesis, reactive side chains of the amino acids were also modified with the appropriate protecting groups. Upon completion of the chain assembly, the peptides were cleaved from the resin by simultaneous removal of the side chain-protecting groups. The cleavage of the synthesized peptides from the resin was performed using trifluoroacetic acid (TFA) with appropriate scavengers (phenol, triisopropylsilane (TIS), water). Subsequently, the compounds were precipitated from diethyl ether and dried under vacuum. The probes were further characterized by the high resolution electrospray ionization mass spectrometry and its purity was assessed by the preparative HPLC. The general scheme of Fmoc solid phase synthesis is presented in Figure 1.
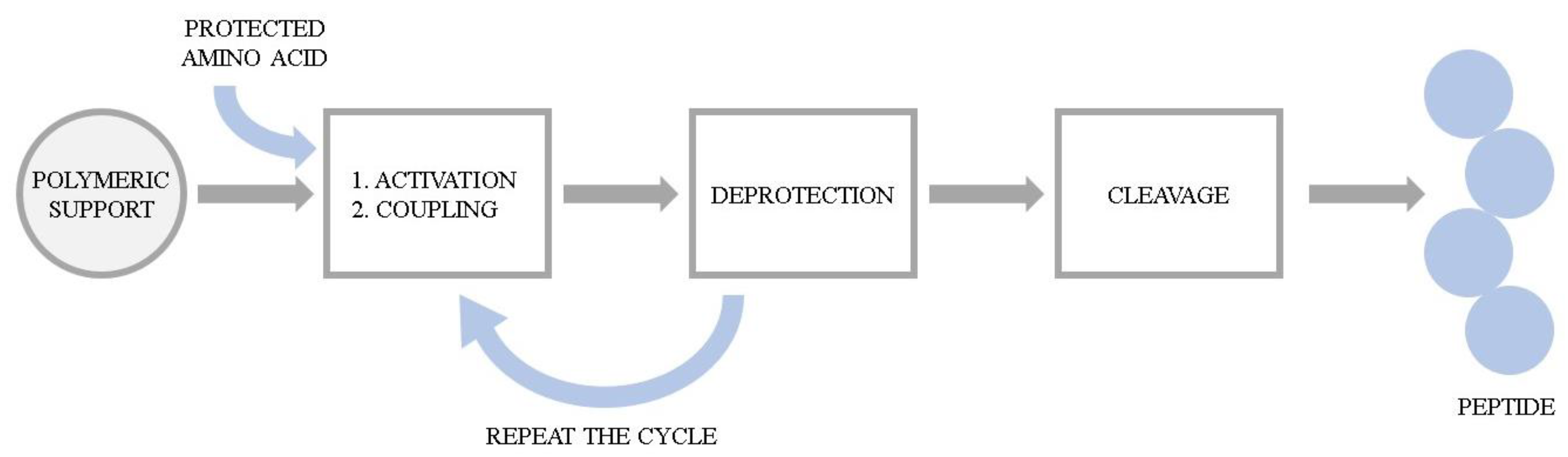 Figure 1. The general scheme of Fmoc solid phase peptide synthesis.
Figure 1. The general scheme of Fmoc solid phase peptide synthesis.To obtain the azobenzene-containing hemorphins, a solid-phase dimerization strategy was applied. This idea is based on the condensation of two units of the heptapeptide (VVYPWTQ) with the azobenzene moiety directly on the resin. Mainly, after formation of the heptapeptide (VVYPWTQ), the azobenzene-4,4′-dicarboxylic acid was coupled using TBTU/DIPEA and the coupling dimerization reaction was performed. Peptides were further cleaved from the resin, purified and analyzed according to the methods described above.
In the first set of experiments, C-amide peptide derivatives were prepared by incorporating non-proteinogenic/natural amino acids and azobenzene moiety in VVYPWTQ-NH2 (VV-hemorphin-5) and VVYPWTQRF-NH2 (VV-hemorphin-7) sequences. These analogues were further tested for visceral nociception using the writhing test in mice. The anticonvulsant activity of peptide derivatives was also evaluated in the three acute seizure tests; the maximal electroshock, the 6 Hz psychomotor seizure test, and the timed intravenous pentylenetetrazole infusion test. Concerning antinociceptive potency, the replacement of Gln position by Dap (2,3-diaminopropanoic acid) in VV-hemorphin-5 led to a derivative exhibiting the strongest analgesic effect among all peptides studied. It is noteworthy that, when isoleucine or 2-aminoisobutyric acid was added at position 1, a decrease in biological activity has been observed. Incorporation of Orn (2,5-diaminopentanoic acid) at Gln position of VV-hemorphin-5 and Dab (2,5-diaminobutanoic acid) moiety at Gln position of VV-hemorphin-7 peptide, respectively led to the analogues posessing good efficacy against the three seizure models [25][24][26][27][28].
VV-hemorphin-5 analogues modified at N-terminal with aminophosphonate group were characterized as potential anticonvulsants. Incorporation of (dimethoxyphosphyl)methyl)-leucine) moiety at two N-terminal Val positions allowed the most active derivative to be obtained [27]. To find an explanation of the possible molecular mechanism of action of the peptides with anticonvulsive properties, docking studies on delta- (DOR) and kappa- (KOR) opioid receptors have been elucidated. The results confirmed experimental data and suggested that the mechanism of the action of these peptides mostly involves activation of KORs.
Reports describing strong anticonvulsive and antinociceptive activities of several adamantane-modified molecules prompted Todorov’s group to perform the research in this direction. The new active analog of hemorphin-4 containing both adamantane and cyclohexane moieties have been synthesized [28]. Docking studies were performed using a model involving insulin-regulated aminopeptidase (IRAP), indicating that the obtained analogue is the most potent among tested inhibitors, and its biological activity has been further confirmed in the in vivo experiments.
Overall, design and synthesis of hemorphin analogues has led to significant changes both in the peptides′ activity and affinity towards opioids receptors or IRAP enzyme. Importantly, not only the position, but also the nature of the incorporated group had an impact on this phenomenon. The obtained data provide the basis for anticipation that the described compounds may be considered as a promising template for future design of the more active analogues, as well as in studies of other models of epilepsy [25][24][26][27][28][29].
Altogether, studies indicate that hemorphins comprise promising candidates for the therapy of pain, anxiety and depression or even hyperalgesia induced by ethanol withdrawal. However, the involvement of hemorphins in various pathological and physiological conditions requires future research.
4. Conclusions
Today, hemorphins find their application as therapeutic agents or as biomarkers in various scientific disciplines, from neuropharmacology (pain, schizophrenia, depression, Alzheimer’s disease, and addiction treatment), to diagnostics (biomarkers of various disorders e.g., vascular and renal disease, cancer, inflammation), and finally, their pharmacological potential as future drugs. The presented studies on LVV-H7 distribution indicate a substantial role of opioid peptides in various pathological and physiological processes; however, the concentration of LVV-H7 and the conditions of their release form hemoglobin chains is not fully understood.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26133879
References
- Glämsta, E.L.; Marklund, A.; Hellman, U.; Wernstedt, C.; Terenius, L.; Nyberg, F. Isolation and characterization of a hemoglobin-derived opioid peptide from the human pituitary gland. Regul. Pept. 1991, 34, 169–179.
- Barkhudaryan, N.; Oberthuer, W.; Lottspeich, F.; Galoyan, A. Structure of hypothalamic coronaro-constrictory peptide factors. Neurochem. Res. 1992, 17, 1217–1221.
- Karelin, A.A.; Philippova, M.M.; Karelina, E.V.; Ivanov, V.T. Isolation of endogenous hemorphin-related hemoglobin fragments from bovine brain. Biochem. Biophys. Res. Commun. 1994, 202, 410–415.
- Cerpa-Poljak, A.; Lahnstein, J.; Mason, K.E.; Smythe, G.A.; Duncan, M.W. Mass spectrometric identification and quantification of hemorphins extracted from human adrenal and pheochromocytoma tissue. J. Neurochem. 1997, 68, 1712–1719.
- Yatskin, O.N.; Philippova, M.M.; Blishchenko, E.; Karelin, A.A.; Ivanov, V.T. Lvv- and vv-hemorphins: Comparative levels in rat tissues. FEBS Lett. 1998, 428, 286–290.
- Glämsta, E.L.; Meyerson, B.; Silberring, J.; Terenius, L.; Nyberg, F. Isolation of a hemoglobin-derived opioid peptide from cerebrospinal fluid of patients with cerebrovascular bleedings. Biochem. Biophys. Res. Commun. 1992, 184, 1060–1066.
- Nyberg, F.; Sanderson, K.; Glämsta, E.L. The hemorphins: A new class of opioid peptides derived from the blood protein hemoglobin. Biopolymers 1997, 43, 147–156.
- Brantl, V.; Gramsch, C.; Lottspeich, F.; Mertz, R.; Jaeger, K.H.; Herz, A. Novel opioid peptides derived from hemoglobin: Hemorphins. Eur. J. Pharmacol. 1986, 125, 309–310.
- Fruitier, I.; Garreau, I.; Lacroix, A.; Cupo, A.; Piot, J.M. Proteolytic degradation of hemoglobin by endogenous lysosomal proteases gives rise to bioactive peptides: Hemorphins. FEBS Lett. 1999, 447, 81–86.
- Piot, J.M.; Zhao, Q.; Guillochon, D.; Ricart, G.; Thomas, D. Isolation and characterization of two opioid peptides from a bovine hemoglobin peptic hydrolysate. Biochem. Biophys. Res. Commun. 1992, 189, 101–110.
- Jinsmaa, Y.; Yoshikawa, M. Release of hemorphin-5 from human hemoglobin by pancreatic elastase. Biosci. Biotechnol. Biochem. 2002, 66, 1130–1132.
- Fruitier, I.; Garreau, I.; Piot, J.M. Cathepsin D is a good candidate for the specific release of a stable hemorphin from hemoglobin in vivo: VV-hemorphin-7. Biochem. Biophys. Res. Commun. 1998, 246, 719–724.
- Garreau, I.; Cucumel, K.; Dagouassat, N.; Zhao, Q.; Cupo, A.; Piot, J.M. Hemorphin peptides are released from hemoglobin by cathepsin d. Radioimmunoassay against the c-part of v-v-hemorphin-7: An alternative assay for the cathepsin d activity. Peptides 1997, 18, 293–300.
- Zadina, J.E.; Kastin, A.J.; Kersh, D.; Wyatt, A. Tyr-mif-1 and hemorphin can act as opiate agonists as well as antagonists in the guinea pig ileum. Life Sci. 1992, 51, 869–885.
- Silberring, J.; Nyberg, F. Rapid analysis of endogenous lvv-hemorphin-7 in cerebrospinal fluid by size-exclusion chromatography and electrospray ionization mass spectrometry. J. Chromatogr. A 1997, 777, 41–45.
- Mak, P.; Wójcik, K.; Wicherek, L.; Suder, P.; Dubin, A. Antibacterial hemoglobin peptides in human menstrual blood. Peptides 2004, 25, 1839–1847.
- Lammerich, H.P.; Busmann, A.; Kutzleb, C.; Wendland, M.; Seiler, P.; Berger, C.; Eickelmann, P.; Meyer, M.; Forssmann, W.G.; Maronde, E. Identification and functional characterization of hemorphins vv-h-7 and lvv-h-7 as low-affinity agonists for the orphan bombesin receptor subtype 3. Br. J. Pharmacol. 2003, 138, 1431–1440.
- Barkhudaryan, N. In vivo microdialysis is a tool to study the mechanism of interaction between lvv-hemorphin-7 and brain serotonergic system. Biotechnol. Health Yerevan 2005, 32–42.
- Ivanov, V.T.; Karelin, A.A.; Philippova, M.M.; Nazimov, I.V.; Pletnev, V.Z. Hemoglobin as a source of endogenous bioactive peptides: The concept of tissue-specific peptide pool. Biopolymers 1997, 43, 171–188.
- Sanderson, K.; Nyberg, F.; Khalil, Z. Modulation of peripheral inflammation by locally administered hemorphin-7. Inflamm. Res. 1998, 47, 49–55.
- Moisan, S.; Harvey, N.; Beaudry, G.; Forzani, P.; Burhop, K.E.; Drapeau, G.; Rioux, F. Structural requirements and mechanism of the pressor activity of leu-val-val-hemorphin-7, a fragment of hemoglobin beta-chain in rats. Peptides 1998, 19, 119–131.
- Darvesh, A.S.; Carroll, R.T.; Geldenhuys, W.J.; Gudelsky, G.A.; Klein, J.; Meshul, C.K.; Van der Schyf, C.J. In vivo brain microdialysis: Advances in neuropsychopharmacology and drug discovery. Expert Opin. Drug Discov. 2011, 6, 109–127.
- Jessop, D.S. Neuropeptides in the immune system: Mediators of stress and inflammation. In Handbook of Neurochemistry and Molecular Biology; Edition: Neuroimmunology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 19–35.
- Todorov, P.; Peneva, P.; Pechlivanova, D.; Georgieva, S.; Dzhambazova, E. Synthesis, characterization and nociceptive screening of new vv-hemorphin-5 analogues. Bioorg. Med. Chem. Lett. 2018, 28, 3073–3079.
- Todorov, P.T.; Peneva, P.N.; Georgieva, S.I.; Tchekalarova, J.; Vitkova, V.; Antonova, K.; Georgiev, A. Synthesis, characterization and anticonvulsant activity of new azobenzene-containing vv-hemorphin-5 bio photoswitch. Amino. Acids 2019, 51, 549–563.
- Todorov, P.; Rangelov, M.; Peneva, P.; Todorova, N.; Tchekalarova, J. Anticonvulsant evaluation and docking analysis of vv-hemorphin-5 analogues. Drug Dev. Res. 2019, 80, 425–437.
- Todorov, P.; Peneva, P.; Tchekalarova, J.; Georgieva, S. Potential anticonvulsant activity of novel vv-hemorphin-7 analogues containing unnatural amino acids: Synthesis and characterization. Amino Acids 2020, 52, 567–585.
- Todorov, P.; Peneva, P.; Tchekalarova, J.; Georgieva, S.; Rangelov, M.; Todorova, N. Structure-activity relationship study on new hemorphin-4 analogues containing steric restricted amino acids moiety for evaluation of their anticonvulsant activity. Amino Acids 2020, 52, 1375–1390.
- Todorov, P.; Peneva, P.; Tchekalarova, J.; Rangelov, M.; Georgieva, S.; Todorova, N. Synthesis, characterization and anticonvulsant activity of new series of n-modified analogues of vv-hemorphin-5 with aminophosphonate moiety. Amino Acids 2019, 51, 1527–1545.
This entry is offline, you can click here to edit this entry!