Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pathology
EWSR1 belongs to the FET family of RNA-binding proteins including also Fused in Sarcoma (FUS), and TATA-box binding protein Associated Factor 15 (TAF15).
- EWSR1
- soft tissue tumors
- bone tumors
- pathology
- molecular
1. Introduction
Ewing sarcoma was molecularly defined by Delattre et al. in 1992 upon the identification of the Ewing sarcoma breakpoint region 1 (EWSR1) located on chromosome 22q12.2 and the term for this gene was coined [1]. EWSR1 is a multifunctional protein ubiquitously expressed in most cell types, indicating diverse roles in physiological cellular processes, including organ development and aging. Genetic and epigenetic pathways are modulated by EWSR1 but the exact mechanisms are still poorly understood [2].
EWSR1 belongs to the FET (also known as TET) family of RNA-binding proteins that also includes Fused in Sarcoma (FUS), and TATA-box binding protein Associated Factor 15 (TAF15) [2]. As a consequence of the multifunctional role of EWSR1 leading to a high frequency of transcription of the chromosomal region where the gene is located, EWSR1 is exposed to aberrations such as rearrangements. Consecutive binding to other genes leads to chimeric proteins inducing oncogenesis. These various somatic genetic rearrangements involving EWSR1 result in a fusion of its N-terminal coding region to the C-terminal DNA binding domain of one of several transcription factors. They are reported to act as aberrant transcription factors with the N-terminal domain of EWSR1 as a strong transactivator. The other TET family members are homologous and are involved in strikingly similar translocation events giving rise to the production of structurally similar oncoproteins [3][4].
With the advent of widely used modern molecular techniques during the last decades, it has become obvious that EWSR1 is involved in development of diverse benign and malignant tumors with mesenchymal, neuroectodermal, and epithelial/myoepithelial features [5]. As oncogenic transformation mediated by EWSR1-fusion proteins leads to such diverse tumor types, there must be a selection on a multipotent stem cell level [2].
2. Ewing Sarcoma
Arthur Purdy Stout and James Ewing were the first to describe this aggressive small, blue round-cell entity in 1918 and 1921, respectively [6][7][8]. Later on, the chromosomal translocation (11;22) was found by Aurias et al. and Turc-Carel et al. in 1983, the second breakthrough of translocation/fusion-gene associated sarcomas following alveolar rhabdomyosarcoma (ARMS) [9][10][11]. Subsequently, the fusion gene has been detected as mentioned in the introduction [1], being the genetic hallmark by an otherwise aspecific small blue, round-cell tumor.
Ewing sarcoma, the prototypic round-cell sarcoma, is relatively common in comparison to other small blue round-cell sarcomas. It arises in soft tissue and bone of children, adolescents, and young adults. Exceptionally, older patients are affected. The mean age is in the second to third decade. White males have the highest incidence and black females the lowest due to ethnic genetic preposition differences. Tumors can originate anywhere in the body, and around 80% of the neoplasms arise in the bone with preference sites in decreasing order of frequency: lower extremities, pelvis, upper extremities, ribs, spine, and craniofacial. Distribution in the soft tissue is extremities, chest wall, retroperitoneum, paravertebral, pelvis, and head and neck. Visceral organs, skin, and epidural spaces are rarely involved [12][13]. The origin of the peripheral nerve as reported by Stout in 1918 can clinically be confused with malignant peripheral nerve sheath tumor [7].
Macroscopically, these infiltrative lesions are (multi)nodular), fleshy, and often necrotic. A pseudocapsule can be present in soft tissue neoplasms. Post-therapy specimens show fibrosis, necrosis, and hemorrhage, often without visible tumor [12][13].
Histologically, Ewing sarcoma is composed of cellular sheets of relatively featureless small cells with round dark nuclei and inconspicuous cytoplasm (Figure 1). In some cases, cells are larger displaying more nuclear variability. The cytoplasm can appear clear due to retraction artefacts. Homer-Wright rosettes may be numerous in a subset of cases initially called peripheral primitive neuroectodermal tumors [6][13]. Adamantinoma-like Ewing sarcoma shows more cohesive sheets and nests of cells with peripheral palisading, prominent desmoplastic stroma with production of hyaline membrane collagen, presence of keratin pearl formation, and comedo-like necrosis. These lesions are predestinated for misinterpretation as carcinoma, since keratins, including high molecular keratins, p40, and p63, are commonly positive [6][14].
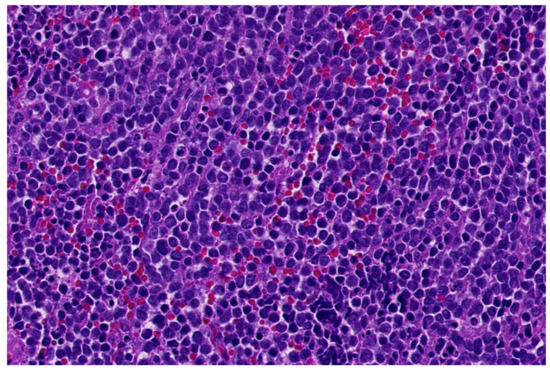
Figure 1. Classical morphology of Ewing sarcoma (HE; 40× magnification).
Immunohistochemically, CD99 is specific in its distinct staining pattern of the cell-membrane. Nuclear FLI and ERG expression is commonly observed in the cases with corresponding fusion genes. Neuroendocrine markers may be expressed. Keratin-expression, often dot-like, was found in 1/3 of the cases; it can be confused with small-cell carcinoma, especially when combined with the expression of p40 and p63 [6][14]. This is of particular importance in the head and neck area [14]. Expression of NKX2-2 in Ewing sarcoma seems to be highly sensitive, with imperfect specificity in comparison to other small, blue round-cell tumors [15][16][17][18][19]. Expression of desmin is reported in a few cases, and can be confused with ARMS or desmoplastic small round-cell tumor (DSRCT) [6][20].
Ewing sarcoma is genetically characterized by binding of EWSR1 or other members of the TET/FET family to members of the ETS family [5]. Approximately 85–90% of the Ewing’s sarcomas display the translocation t(11;22)(q24;q12) resulting in the EWS/FLI1 fusion gene, and approximately 5–10% harbor a EWSR1-ERG fusion gene [6]. The remaining cases show rare gene partners, such as ETV1, ETV4, and FEV, and EWSR1 can be substituted by FUS [21].
Although prognosis has improved markedly for patients with primary disease (5-year survival rate around 65%), presence of metastatic disease at time of diagnosis or early relapse leads to an adverse prognosis (5-year survival rate around 25–30%), with adequate surgical resection, aggressive multimodal chemotherapy, and adjuvant local radiotherapy being the optimal treatments.
Differential diagnoses are listed in Table 1.
Table 1. Differential diagnoses of Ewing sarcoma.
| Entity | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| CIC-sarcoma | Sheets of undifferentiated round/spindle/epitheloid cells; mild nuclear pleomorphism; and necrosis | CD99 (mostly patchy), WT1, ETV4, DUX4, and NUT (CIC-NUTM1) | CIC-DUX4/FOXO4/LEUTX/NUTM1/2A fusions |
| BCOR-sarcoma | Sheets/nests/short fascicles of uniform; bland round-oval-spindle cells; rich capillary network; and myxoid matrix (variable) | BCOR, SATB2, cyclin D1, TLE1, CCNB3 (BCOR-CCNB3), and CD99 (50%) | BCOR-CCNB3/MAML3/ZC3H7B, KMT2D); BCOR ITD*; and YWHAE-NUTM2B; *ITD, internal tandem duplication |
| EWSR1-nonETS round-cell sarcomas | Cords/nests/pseudoacinar pattern of round-spindle cells; bland-pleomorphic spectrum; and fibro-/myxohyaline stroma | CD99, NKX2.2, and CKAE1/3 (focal, dot-like) | EWSR1/FUS-NFATc2 |
| Diverse morphology: round-spindle cells; fibrous stroma | Co-expression of myogenic markers (desmin/myogenin/MyoD1), neurogenic markers (S100/SOX10/MITF/GFAP) and keratins (AE1/3) | EWSR1-PATZ1 or EWSR1-VEZF1 | |
| Desmoplastic small round-cell tumor | Sheets/nests/cords of uniform; bland round cells; and desmoplastic stroma | Desmin (dot-like), keratin, EMA, and WT1 (C-terminus) | EWSR1-WT1 |
| Lymphoblastic lymphoma | Small-medium blastoid cells; minimal cytoplasm | CD99, TdT, CD45, CD34, CD1a, and B- and T-cel markers | Diverse |
| Small-cell carcinoma | Small-medium round-oval cells; salt and pepper chromatin; indistinct nucleoli; molding; and apoptosis | Keratins, CD56, synaptophysin, chromogranin, and TTF1 |
Diverse; TP53, PTEN mutations; RB1, 3p loss; and MYC amplification |
| NUT carcinoma | Poorly cohesive sheets of primitive/basaloid cells; abrupt keratinization; and coagulative necrosis | CK5/6, P40, P63, and NUT | NUT-BRD3/BRD4/NSD3/CIC/BCORL1/MGA/MXD4 |
| Myoepithelial carcinoma | Solid sheets/nests of cell with high nuclear grade or undifferentiated round-cell morphology; facultatively glandular component; necrosis; and high mitotic count | Pankeratins, S100, EMA, GFAP, SOX10, P63, SMA, calponin, desmin (focal); and INI1 loss (subset) | EWSR1 rearrangements (various fusion partners); PLAG1 rearrangements (mixed tumors) |
| ARMS | Nests with central discohesion-solid nests; monomorphic primitive round cells; and multinucleated wreath-like giant cells | Desmin, myogenin (strong, diffuse), MyoD1, keratin, neuro-endocrine markers (CD56, synaptophysin, and chromogranin) | PAX3/PAX7-FOXO1 |
| Sinonasal glomangiopericytoma | Solid-fascicular pattern; spindle-round cells with minimal atypia; arranged around staghorn vessels; and perivascular hyalinization | Beta-catenin (nuclear), SMA | CTNNB1 mutations |
| Glomus tumor | Solid-nested pattern; small, uniform round cells with round nucleus, amphophilic-slightly eosinophilic cytoplasm and sharply defined cell borders; and variable vascular pattern | SMA with membranous accentuation, caldesmon, and collagen IV | MIR143-NOTCH1/2/3, and BRAF/KRAS mutations |
| Rhabdoid tumor | Solid pattern; rounded-polygonal cells with vesicular nuclei and prominent nucleoli; and eosinophil hyaline-like cytoplasmic inclusions | Diverse; keratins, EMA, CD99, synaptophysin, SALL4, glypican-3, and INI1 loss | SMARCB1 biallelic loss, SMARCB1 or SMARCA4 (germline) mutations |
| Mesenchymal chondrosarcoma | Biphasic: poorly differentiated round cells and islands of hyaline cartilage; staghorn-like vessels | S100, CD99, SOX9, EMA, desmin, myogenin, and MyoD1 | HEY1-NCOA2 |
| Synovial sarcoma with round-cell features | Fascicles or sheets of small round hyperchromatic cells; high N/C ratio; staghorn vessels; necrosis; and thin fibrovascular septa | CD99, BCL2, CD56, TLE1, S100 (focal), EMA, and keratins (variable) | SS18-SSX1/2/4 |
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics11061093
References
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165.
- Lee, J.; Nguyen, P.T.; Shim, H.S.; Hyeon, S.J.; Im, H.; Choi, M.H.; Chung, S.; Kowall, N.W.; Lee, S.B.; Ryu, H. EWSR1, a multifunctional protein, regulates cellular function and aging via genetic and epigenetic pathways. Biochim. Biophys. Acta Mol. Basis Dis 2019, 1865, 1938–1945.
- Bertolotti, A.; Bell, B.; Tora, L. The N-terminal domain of human TAFII68 displays transactivation and oncogenic properties. Oncogene 1999, 18, 8000–8010.
- Lindén, M.; Thomsen, C.; Grundevik, P.; Jonasson, E.; Andersson, D.; Runnberg, R.; Dolatabadi, S.; Vannas, C.; Luna Santamarίa, M.; Fagman, H.; et al. FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Rep. 2019, 20.
- W.H.O. Classification of Soft Tissue and Bone Tumours; International Agency for Research on Cancer: Lyon, France, 2020.
- Folpe, A.L.; Goldblum, J.R.; Rubin, B.P.; Shehata, B.M.; Liu, W.; Dei Tos, A.P.; Weiss, S.W. Morphologic and immunophenotypic diversity in Ewing family tumors: A study of 66 genetically confirmed cases. Am. J. Surg. Pathol. 2005, 29, 1025–1033.
- Stout, A.P. A tumor of ulnar nerve. Proc. NY Pathol. Soc. 1918, 18, 2–12. (In Japanese)
- Ewing, J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J. Clin. 1972, 22, 95–98.
- Seidal, T.; Mark, J.; Hagmar, B.; Angervall, L. Alveolar rhabdomyosarcoma: A cytogenetic and correlated cytological and histological study. Acta Pathol. Microbiol. Immunol. Scand. A 1982, 90, 345–354.
- Aurias, A.; Rimbaut, C.; Buffe, D.; Dubousset, J.; Mazabraud, A. Translocation of chromosome 22 in Ewing’s sarcoma. C R Seances Acad Sci. III 1983, 296, 1105–1107.
- Turc-Carel, C. Chromosomal translocations in Ewing’s sarcoma. N. Engl. J. Med. 1983, 309, 496–498.
- Czerniak, B.U. Dorfman and Czerniak’s Bone Tumors; Elsevier Health Sciences: Houston, TX, USA, 2015.
- Miettinen, M.U. Modern Soft Tissue Pathology: Tumors and Non-Neoplastic Conditions; Cambridge University Press: Cambridge, UK, 2010.
- Rooper, L.M.; Bishop, J.A. Soft Tissue Special Issue: Adamantinoma-Like Ewing Sarcoma of the Head and Neck: A Practical Review of a Challenging Emerging Entity. Head Neck Pathol. 2020, 14, 59–69.
- Yoshida, A.; Sekine, S.; Tsuta, K.; Fukayama, M.; Furuta, K.; Tsuda, H. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am. J. Surg. Pathol. 2012, 36, 993–999.
- Shibuya, R.; Matsuyama, A.; Nakamoto, M.; Shiba, E.; Kasai, T.; Hisaoka, M. The combination of CD99 and NKX2.2, a transcriptional target of EWSR1-FLI1, is highly specific for the diagnosis of Ewing sarcoma. Virchows Arch. 2014, 465, 599–605.
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: Imperfect specificity for Ewing sarcoma. Mod. Pathol. 2016, 29, 370–380.
- Russell-Goldman, E.; Hornick, J.L.; Qian, X.; Jo, V.Y. NKX2.2 immunohistochemistry in the distinction of Ewing sarcoma from cytomorphologic mimics: Diagnostic utility and pitfalls. Cancer Cytopathol. 2018, 126, 942–949.
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006, 9, 405–416.
- Barisella, M.; Collini, P.; Orsenigo, M.; Aiello, A.; Paties, C.T.; Dileo, P.; Pilotti, S. Unusual myogenic and melanocytic differentiation of soft tissue pPNETs: An immunohistochemical and molecular study of 3 cases. Am. J. Surg. Pathol. 2010, 34, 1002–1006.
- Thway, K.; Fisher, C. Mesenchymal Tumors with EWSR1 Gene Rearrangements. Surg. Pathol. Clin. 2019, 12, 165–190.
This entry is offline, you can click here to edit this entry!