Cyclotides are a novel class of micro-proteins (≈30–40 residues long) with a unique topology containing a head-to-tail cyclized backbone structure further stabilized by three disulfide bonds that form a cystine knot. This unique molecular framework makes them exceptionally stable to physical, chemical, and biological degradation compared to linear peptides of similar size. The cyclotides are also highly tolerant to sequence variability, aside from the conserved residues forming the cystine knot, and are orally bioavailable and able to cross cellular membranes to modulate intracellular protein–protein interactions (PPIs), both in vitro and in vivo. These unique properties make them ideal scaffolds for many biotechnological applications, including drug discovery.
1. Introduction
The selective disruption of pharmacologically relevant protein–protein interactions (PPIs) still remains a very challenging task [
1,
2,
3]. This is mostly due to the relatively large and relatively flat nature of the binding surfaces involved in most PPIs. The most challenging of the molecular targets are in fact those involving intracellular PPIs, which, in addition, require the therapeutic agent to be able to cross the cell membrane in an efficient manner [
4,
5].
In general, we can consider two major structural types of therapeutic agents, small molecules and protein-based compounds, with the later also known as biologicals. Small molecules, as their name indicates, are small in molecular size (≤100 atoms) and in general show good pharmacological properties, such as stability and cell permeability. Their intrinsic small size, however, only provides a modest overall surface area available to interact with the target protein, making the identification of small molecules able to efficiently disrupt PPIs quite challenging [
6,
7].
On the other hand, the use of polypeptide-based molecules has been able to provide efficient therapeutic tools to modulate PPIs with high specificity and selectivity [
8]. The use of therapeutic monoclonal antibodies to target extracellular protein receptors is just one example [
9,
10]. Antibodies, however, suffer from clear limitations: they are expensive to produce, cannot be delivered orally, show low tissue penetration, and are unable to reach intracellular targets. These issues have led to the exploration of alternative protein scaffolds as a source for novel protein-based therapeutics [
11,
12,
13,
14,
15,
16].
Special attention has been recently given to the use of highly constrained polypeptides for the development of novel stable polypeptide-based therapeutics [
17,
18,
19]. Cyclotides are a fascinating emerging family of large plant-derived backbone-cyclized polypeptides (≈30–40 amino acids long) containing a 3 disulfide-stabilized core characterized by an unusual knotted structure (
Figure 1) [
20]. This unusual topology confers the cyclotide scaffold with unique characteristics that make them ideal for drug development (see recent reviews [
21,
22,
23]).
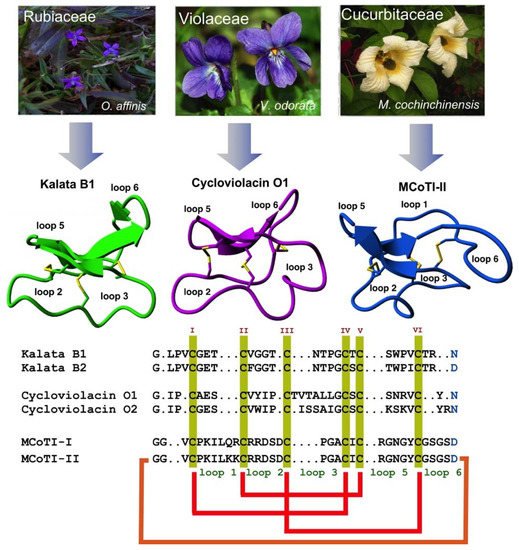
Figure 1. Biological origin, structures, and sequence alignment of different cyclotides belonging to the Möbius (kalata B1, pdb: 1NB1) [
24], bracelet (cycloviolacin O1, pdb: 1NBJ) [
24], and trypsin inhibitor (MCoTI-II, pdb: 1IB9) [
25] subfamilies. These three naturally-occurring cyclotides were isolated from
O. affinis (
Rubiaceae family),
Viola odorata (
Violaceae family), and
M. cochinchinensis (
Cucurbitaceae family). The six Cys residues are labeled with roman numerals, whereas loops connecting the different Cys residues are designated with Arabic numerals. Conserved Cys and Asp/Asn (required for backbone cyclization in nature) residues are marked in yellow and light blue, respectively. Disulfide connectivities and backbone-cyclization are shown in red and orange, respectively. Molecular graphics were created using Yasara (
www.yasara.org). Figure adapted from references [
17,
23].
Cyclotides are remarkably stable to thermal and chemical denaturation and biological degradation by proteolytic enzymes [
26]. They can be easily accessible by chemical synthesis due to their relative small size and can be also recombinantly produced using standard expression vectors in different types of cells (see a recent review on the production of cyclotides [
27]). Some cyclotides have been shown to be able to cross the cellular membranes of mammalian cells [
28,
29] to modulate intracellular PPIs, both in vitro and in vivo [
5]. Even more exciting, cyclotides have shown to have biological activity when dosed orally [
26,
30,
31].
The naturally-occurring cyclotide kalata B1, which was the first cyclotide to be discovered in plants, was used as an orally effective uterotonic [
26] and several other kalata B1-based cyclotides have also been shown to be orally bioavailable [
30,
31]. These unusual characteristics for a polypeptide-based molecular scaffold make the cyclotide molecular framework an ideal substrate for molecular engineering and evolution strategies for the production of novel peptide-based diagnostic, therapeutic, and research tools. This article is meant to provide a brief overview of their most relevant properties and their potential to be used as a molecular scaffold for the development of peptide-based therapeutic agents.
2. Structure
All naturally-occurring cyclotides are backbone-cyclized and contain between 27 to 37 amino acids, of which six are Cys residues. The six Cys residues form three disulfide bonds adopt a cystine-knot topology, with disulfides Cys
I-Cys
IV and Cys
II-Cys
V forming a ladder arrangement and disulfide Cys
III-Cys
VI running through it (
Figure 1). This highly interlocked cyclic cystine knot (CCK) motif makes the backbone of cyclotides very rigid and compact [
32], which is responsible for their high stability to thermal, chemical, and proteolytical degradation [
33,
34]. This is highlighted in the case of the first cyclotide to be isolated, kalata B1, which was identified in the late 1960s by Gran when studying an indigenous traditional medical remedy in central Africa that was used to facilitate childbirth in pregnant women [
35]. This traditional remedy used a tea obtained from the plant
Oldelandia affinis from the
Rubiaceae family [
36]. The fact that the cyclotide kalata B1 was able to remain folded and biologically active even after being extracted by boiling water to produce a medicinal tea with uterotonic properties shows the remarkable stability of the cyclotide scaffold.
Cyclotides can be classified into three subfamilies, the Möbius, bracelet, and trypsin inhibitor cyclotide subfamilies [
37]. All the subfamilies share the CCK topology, however, the loop composition, size, and sequence can be different among the members of the three subfamilies. Cyclotides from the Möbius sub-family, such as kalata B1, have a
cis-Pro bond at loop 5 formed by a cis tryptophan–proline bond resulting in an 180° twist of the peptide backbone, while bracelet cyclotides do not have it [
24].
Bracelet cyclotides are the most abundant in nature, making up ≈66% of the all the sequenced cyclotides known thus far [
38]. These cyclotides are more structurally different and slightly larger in size than those from the Möbius subfamily. Bracelet cyclotides, on the other hand, are more difficult to fold in vitro than either Möbius or trypsin inhibitor cyclotides, making them more challenging to produce by using standard peptide synthesis protocols [
39]. For that reason, cyclotides from the bracelet subfamily are much less used as molecular scaffolds for the production of cyclotides with novel biological activities.
The third subfamily of cyclotides, the trypsin inhibitor subfamily, contains only a small number of cyclotides isolated from the seeds of several
Momordica spp plants (
Cucurbitaceae family) [
40,
41,
42]. Cyclotides from this subfamily are very potent trypsin inhibitors (
Ki ≈ 20 pM) [
43] that do not share significant sequence homology with cyclotides from the other two subfamilies beyond the CCK topology common to all cyclotides (
Figure 1). Cyclotides from the trypsin subfamily show high sequence homology with cystine knot squash trypsin inhibitors and sometimes are referred to as cyclic knottins [
44].
More recently, a new type of cyclotides with high content in positively-charged Lys residues haven also been isolated from two species of Australasian plants from the
Violaceae family [
45]. Unfortunately, there is not yet any information on their chemical synthesis, making it difficult to evaluate their real potential for being used as molecular frameworks in the design of novel peptide-based therapeutics.
3. Biosynthesis
Naturally-occurring cyclotides are produced by enzymatic processing from ribosomally produced precursor proteins [
37]. Many cyclotides have dedicated genes encoding multiple copies of the same cyclotide or mixtures of different cyclotide sequences [
46]. Not surprisingly, the first dedicated genes encoding cyclotide precursor proteins were found in the cyclotide producing plant
Oldelandia affinis (
Rubiaceae family), which is the source of the cyclotide kalata B1 [
47,
48]. Similar genes have also been found in other plants from the
Violaceae and
Rubiaceae [
49], and more recently also in plants from the
Solanaceae,
Fabaceae, and
Cucurbitaceae families [
20,
41,
49,
50,
51]. Some of these new genes provide novel architectures for the corresponding cyclotide protein precursor, showing the high diversity used by nature to generate cyclotides.
Although the post-translational modifications required for the biosynthesis of cyclotides in nature have not been fully characterized yet [
49,
52], recent studies have determined that asparaginyl endopeptidase (AEP)-like ligases are involved in the C-terminal cleavage and backbone-cyclization of the linear cyclotide precursor [
53,
54]. For example, the co-expression of a cyclizing AEP-like ligase with a cyclotide precursor protein in non-cyclotide producing plants significantly improves the cyclization efficiency of the corresponding cyclotide linear precursor [
55]. These AEP-like ligases have been shown to be able to backbone-cyclize different linear peptides in vitro, including linear cyclotide precursors and even peptides containing D-amino acids [
53,
55,
56,
57,
58,
59].
Despite the increasing understanding of how cyclotides are produced in plants, there is still not too much known about the N-terminal cleavage process and the corresponding associated protease. Complete understanding on how the cyclotide linear precursors are processed and identification of all the players involved should facilitate the engineering of genetically-modified organisms for the inexpensive bioproduction of cyclotides [
60,
61].
4. Chemical Synthesis
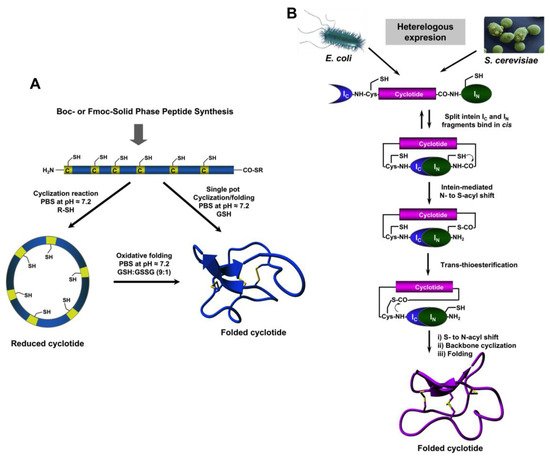
The relatively small size of cyclotides makes the synthesis of the corresponding linear precursors by chemical methods possible, using solid-phase peptide synthesis (SPPS) [
27]. Backbone cyclization of the linear precursor can be easily accomplished in aqueous buffers at pH ≈7 using an intramolecular version of native chemical ligation (
Figure 2A). The required peptide α-thioester can be readily generated using standard solid-phase peptide synthesis methods by either Boc- or Fmoc-based chemistry [
27]. The corresponding linear precursor can be cyclized and oxidatively folded sequentially. A very convenient approach to generate chemically-produced cyclotides involves carrying out the cyclization and folding steps in a “single pot” reaction by using glutathione (GSH) as a thiol additive [
62]. This approach has successfully been used to chemically generate many native and engineered cyclotides [
5,
62,
63,
64], as well as other disulfide-contained backbone-cyclized polypeptides [
65,
66].
Figure 2. Different available approaches for the production of cyclotides. (
A) Chemical synthesis of cyclotides by making use of an intramolecular version of native chemical ligation. This approach requires the generation of a linear precursor polypeptide bearing an N-terminal Cys residue and an α-thioester moiety at the C-terminus. The linear precursor can be first cyclized under reductive conditions and then folded using a proper redox buffer, for example using reduced and oxidized glutathione (GSH) [
27]. The cyclization and oxidative folding can be also efficiently accomplished in a “single pot” reaction when the cyclization is carried out in the presence of reduced GSH as the thiol cofactor [
27]. (
B) Recombinant expression of cyclotides by making use of the protein trans-splicing (PTS) [
73,
74,
75]. This approach has been employed for the generation of several MCoTI-cyclotides, where the native Cys residue located at the beginning of loop 6 was used to facilitate backbone cyclization. This method can be used to produce bioactive cyclotides in either eukaryotic or prokaryotic expression systems [
73,
74,
75]. Figure adapted from a previous study [
23].
Cyclotide linear precursors can be also chemoenzymatically cyclized using AEP-like ligases [
53,
54,
59,
67], which do not require the linear precursor to be natively folded for the cyclization to proceed efficiently [
53]. Naturally occurring trypsin inhibitor cyclotides, such as MCoTI-I/II, can also be produced using the serine protease trypsin [
68]. This is accomplished by producing a folded linear precursor bearing the P1 and P1 residues at the C- and N-termini, respectively. This approach provides a very efficient route for obtaining cyclotides with trypsin inhibitory properties with yields close to 92% for cyclotide MCoTI-II [
68], however the introduction of mutations that affect the binding to the proteolytic enzyme may affect the cyclization yield [
27]. Other proteases, such as the transpeptidase like sortase A (SrtA), have been also employed for the backbone cyclization of the corresponding synthetic linear precursor [
69]. However, this approach, due to the sequence requirements for SrtA to work properly, leaves an extra heptapeptide motif at the cyclization site, which should be taken into consideration when producing bioactive cyclotides.
5. Recombinant Expression
The use of protein splicing units, also called inteins, in either
cis or
trans allows the recombinant production of backbone cyclized polypeptides (for more detailed reviews in this topic see [
27,
70]). Initial attempts for production of cyclotides using heterologous expression systems involved the use of modified inteins for generating α-thioester polypeptides that were then backbone-cyclized using an intramolecular version of native chemical ligation [
71,
72]. The use of intein-mediated protein
trans-splicing (PTS) has been shown to be more effective for the production of naturally-occurring and engineered cyclotides in prokaryotic and eukaryotic expression systems (
Figure 2B) [
73,
74,
75]. In-cell production of folded cyclotides by PTS can reach intracellular concentrations in the range of 2040– μM. This corresponds to ≈ 10 mg of folded cyclotide per 100 g of wet cells in
Escherichia coli expression systems producing cyclotide MCoTI-I [
75]. These values are quite comparable to those obtained when using the cyclotide-producing plant
O. affinis, which produces ≈ 15 mg of cyclotide kalata B1 per 100 g of wet weight when grown in vitro [
76]. Given the fastest growth rate and the simplicity of working with microorganisms such as
E. coli, PTS provides a very attractive alternative for a cost-effective route to produce bioactive cyclotides with therapeutic potential.
In-cell production of cyclotides also opens the exciting possibility for the generation of large genetically-encoded libraries of cyclotides, which can be rapidly screened for the selection of novel sequences able to modulate specific molecular targets [
74]. In addition, having easy access to cyclotides using standard heterologous expression systems facilitates the production of cyclotides labeled with NMR active isotopes, such as
15N and
13C, in a relatively inexpensive fashion [
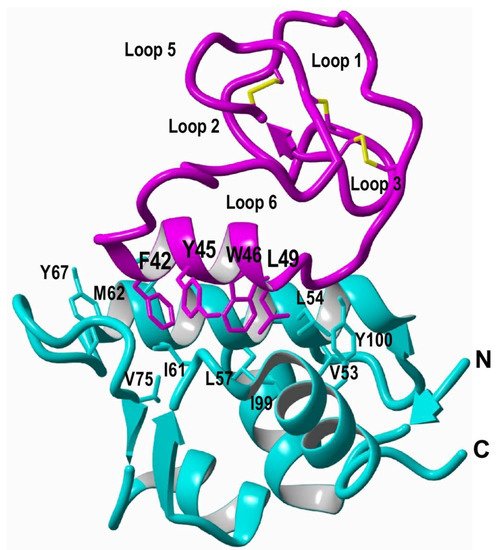
5]. This approach was used to carry out structural studies using heteronuclear NMR on a cyclotide engineered to bind the p53 binding domain of the E3-ligases Hdm2 and HdmX, allowing elucidation of the structure of the bioactive cyclotide bound to its target (
Figure 3) [
5].
Figure 3. Structure of a MCoTI-based cyclotide designed to antagonize an intracellular PPI [
5]. The structure of the engineered cyclotide MCo-PMI (magenta) and its intracellular molecular target, the p53 binding domain of oncogene Hdm2 (blue), were determined in solution by nuclear magnetic resonance (NMR). Cyclotide MCo-PMI binds with low nM affinity to both the p53-binding domains of Hdm2 and HdmX.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines7020031