Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
The deoxyribonuclease 1 (Dnase1) family is a key family of endonucleases that degrades DNA. Loss of Dnase1 family function causes several diseases where the host’s immune system targets the host, such as systemic lupus erythematosus, hypocomplementemic urticarial vasculitis syndrome.
- nuclease
- lupus
- ehrlichiosis
- psoriasis
- HUVS
- NETs
- cell-free DNA
1. History and Overview of the Dnase1 Family
1.1. Discovery of Dnase1 Family Members
Deoxyribonuclease (Dnase) activity was described in bovine organs in the 1800s, and the proteins responsible for this activity have been characterized over the last century. In the early 1900s, digestion of nucleic acids by liver, spleen, pancreas, and other organs was investigated. As reviewed [1], the enzymatic activity was given a succession of names, including desoxyribonuclease, and determined to function optimally at neutral pH [2]. The founding member of the Dnase1 family, Dnase1, was isolated and crystallized in 1950 [1]. Dnase1 activity was generally recognized to require divalent cations (Ca2+ or Mg2+), to act optimally at neutral pH, and to leave 5′ phosphates following DNA cleavage [1,2,3,4]. As reviewed [5], in 1947 a second, “acid DNase” activity was described in mammals [6]. Acid DNase activity had an ubiquitous tissue distribution and showed peak activity at acidic pH [5]. To distinguish the acid DNase from the pancreatic DNase, which was called Dnase I, and later Dnase1, the term Dnase II (later Dnase2) was suggested as an alternative to acid DNase [3]. Dnase2 has no requirement for divalent cations and hydrolyzes double stranded DNA into short oligonucleotides bearing 3′ -phosphate groups [5]. Multiple proteins possess acid and neutral DNase activity. The Dnase II/Dnase2 family now consists of Dnase2a, Dnase2b, and L-Dnase II/SerpinB1 [7]. In the 1990s, three new members of the Dnase I family were discovered, and termed “DNase1-like” proteins: Dnase1L1, Dnase1L2, and Dnase1L3 [8,9,10,11,12,13]. Dnase1-like 1 (Dnase1L1) was discovered in 1995 and first named human Dnase I lysosomal-like (DNL1L) [8]. Dnase1L2 was first described in 1997, during the gene mapping of the Dnase1 [9]. Finally, Dnase1L3 was identified in 1994 from nuclei of rat thymocytes as the third of three nucleases and consequently termed ‘Dnase γ’ [12]. Dnase1L3 was also called novel human Dnase (nhDnase) [13], and liver/spleen DNase (LS-Dnase) [14].
Dnase1 family members often show restricted tissue expression. In humans, Dnase1 is primarily secreted in saliva, intestine, pancreas, kidneys, and urine, but is also present in serum [1,15]. Dnase1L1 is restricted to skeletal muscle and cardiomyocytes [16]. Dnase1L2 is primarily restricted to keratinocytes, and tissues containing them, like the skin [17]. Dnase1L3 is secreted into blood, primarily by myeloid cells [18]. Distinct Dnase1 family members enable tissue-specific DNA degradation, dependent on the function of that tissue. Overall, Dnases are essential for DNA degradation in most animals.
1.2. Evolution of Dnases
Consistent with an essential role for DNA degradation, all Dnase1 family members are widely expressed throughout Metazoa. The phylogenetic distribution of Dnase1 and Dnase1L3 in Animalia predicts that Dnase1L3 is closest to the common ancestral Dnase [19]. Dnase1L3 is present in animals from humans down to corals and sponges, whereas the phylogenetically lowest organism containing both Dnase1 and Dnase1L3 is the placozoan Trichoplax adhaerens [19]. Interestingly, Dnase1 and Dnase1L3 are absent in Protostomia, including arthropods and nematodes [19], though nematodes express Dnase2 [20]. A role for Dnase1 family members in digestion has been proposed for sponges [19]. Both Dnase1 and Dnase1L3 may act in this role in phylogenetically lower organisms, with specialization between these enzymes potentially occurring in higher organisms. For example, the sponge Amphimedon queenslandica secretes Dnase1L3 to digest DNA from its prey [19]. However, some specialization between neutral and acid Dnases is present in sponges. The orange sea sponge Tethya aurantium has both neutral and acid Dnases, which show functional specialization [21]. The Dnase1 activity is present in the cortex, which interacts with the environment, while acid Dnase is expressed in endosomes to facilitate digestion [21]. Overall, the Dnase1 family represents a highly conserved enzyme family for removing DNA.
1.3. Extranuclear DNA Is Inflammatory
Removal of DNA is needed not just for digestion, but also to prevent aberrant inflammation. One key danger signal indicating infection or damage in animals is DNA found outside of its expected locations in the nucleus or mitochondria. Consequently, DNA is a potent inducer of inflammation and autoimmunity. Inflammation and cell death can be triggered after cytoplasmic DNA is sensed by several intracellular DNA sensors, including STimulator of INterferon Genes (STING) and Absent In Melanoma 2 (AIM2) [22]. Toll-like Receptor 9 (TLR9) can sense extracellular DNA [22]. Inflammatory DNA is produced during immunity by the release of Neutrophil Extracellular Traps (NETs). NETs are networks of polynucleosomes released from activated neutrophils that primarily consist of chromatin and histones [23]. These 15–17 nm diameter fibers are lined with antimicrobial enzymes and peptides. NETs are used by neutrophils to kill pathogens. NETs are primarily released by NETosis. NETosis is a form of programmed cell death wherein chromatin, nuclear, and granular contents are decondensed and released in the extracellular area [23]. While effective for clearing pathogens, the failure to degrade NETs causes harmful inflammatory responses [24]. DNA in NETs can be internalized and activate STING, leading to the release of pro-inflammatory interferons [25]. DNA is also antigenic. Anti-DNA antibodies characterize several autoimmune diseases, including Systemic Lupus Erythematosus [26]. Anti-DNA-DNA immune complexes promote Complement deposition, inflammation, and tissue damage [26]. Consequently, DNA degradation after necrosis or programmed cell death is critical to preventing autoimmunity. While the intracellular Dnase2a promotes DNA degradation after phagocytosis, Dnase1 family members degrade DNA prior to phagocytosis [7]. Thus, the Dnase1 family plays a key role in preventing autoimmunity from a host’s own DNA, due to its catalytic ability to degrade DNA.
2. Mechanism of Action for the Dnase1 Family
Mechanism of Dnase1 Family Catalysis
Structural biology has provided key insights into the catalytic mechanism of the Dnase1 family. Improved structures of Dnase1 have helped refine and account for the catalytic mechanism of DNA cleavage used by Dnase1 family members. The Dnase1 family primarily relies on a catalytic His conserved across the broader exonuclease-endonuclease-phosphatase (EEP) superfamily to cleave double-stranded DNA in a divalent cation (Mg2+/Ca2+) dependent manner. The dsDNA substrate of the Dnase1 family is principally B-DNA, longer than three nucleotides, with a slight preference for non-repeating base pairs [27]. However, there is substrate variation within the Dnase1 family, best studied between Dnase1 and Dnase1L3. Dnase1 and Dnase1L3 may cooperate due to differences in substrate activity [28]. Dnase1L3 targets a broader range of substrates, while Dnase1 primarily cleaves naked dsDNA non-specifically. Dnase1 cleaves complexed DNA poorly, though chromatin degradation by Dnase1 is aided by heparin-mediated DNA binding protein displacement [29]. Dnase1L3 cleaves both naked DNA and complexed DNA, including chromatin, nucleosomes, DNA in microparticles/apoptotic blebs, DNA coated with lipids, and single-stranded DNA [11,18,30]. However, Dnase1L3 is inactivated by heparin and can be cleaved by plasmin [29]. After Dnase1 family cleavage, DNA has one backbone ester bond nicked, leaving a 3′ OH and 5′ phosphate [1,4]. The mechanisms proposed for Dnase1-mediated DNA cleavage will provide a foundation for analysis of further enzyme specialization in the Dnase1 family.
Generally, Dnase cleavage must balance the thermodynamically favored hydrolysis of the phosphate backbone of DNA with the kinetically challenging task of nucleophilic attack on a negatively charged phosphate [31]. Three essential features are required to strike this balance and complete phosphate backbone hydrolysis: nucleophilic hydroxide activation, transition state stabilization, and leaving group protonation [32]. Several mechanisms have been proposed to address these three essential features in Dnase1-like family endonuclease reactions [33,34,35,36]. The four mechanisms that have been proposed are the Glu-His-Water, Two Histidine Acid-Base, Double-Divalent Cation, and Single-Cation Carboxylic Acid models (Figure 1).
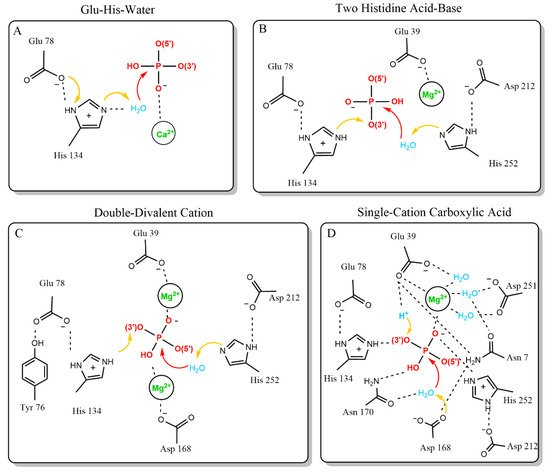
Figure 1. Proposed catalytic mechanisms of Dnase1. The proposed mechanisms are shown in order of first publication date. (A) The first published mechanism, proposed based on the first X-ray structure of [33]. The Glu-His-Water mechanism is similar to serine proteases. (B) The Two-Histidine Acid-Base mechanism is an updated mechanism from the group behind the Glu-His-Water model. It is based on more extensive X-ray structural information [35]. (C) The Double-Divalent Cation mechanism builds on the Two-Histidine Acid-Base model. The Double-Divalent Cation mechanism was based on mutagenesis data [36]. (D) The Single-Cation Carboxylic Acid mechanism reopened the question of Dnase1 family catalysis, based on a more recent substrate-cation crystal structure [34].
The first model for Dnase1 family DNA hydrolysis is the Glu-His-water model. The development of the Glu-His-water model was facilitated by the first crystal structure of Dnase1, and is reminiscent of the serine protease catalytic triad [27,33]. Dnase1 was crystallized in the presence of the calcium ion conjugated nucleotide Ca-pTp. The crystal structure relied on a 4 Å Fourier difference density map for the active site substrate, which did not permit accurate determination of substrate positioning (Figure 1A). However, it could be concluded that a divalent calcium ion permitted phosphate interaction with the otherwise negatively charged active site. The Glu-His-Water model is based on the hydrogen bond distance between the nitrogen on the deprotonated H134 and an opposing carboxyl group in E78. The carboxyl group from E78 polarizes H134, allowing H134 to remove a proton from water, which can then attack the scissile phosphate (Figure 1A). This model accounted for alkylation experiments showing that H134 is a catalytically essential residue [37,38]. The sensitivity of the deprotonated H134 to a lower pH is consistent with the greater activity of Dnase1 at neutral pH. The key weakness in this model is the lack of a proton donor to donate a proton to the 3′ ester bond that completes the reaction.
The Glu-His-Water model was superseded by the Two Histidine Acid-Base catalysis model, which addressed the weakness of the previous model, and extended the catalytic interface of Dnase1. These refinements were made based on the first high-resolution structure of Dnase1 bound to DNA [35]. In this model, Dnase1 interacts with the scissile phosphate via four residues: H134, D168, N170, and H252 [35] (Figure 1B). The updated model changed the role of H134 and assigned new roles to the other amino acids interacting with the scissile phosphate. In contrast to a deprotonated histidine removing a proton from the attacking water, H134 acts as a general acid, and completes the reaction by protonating the 3′ ester. Instead, H252 is the deprotonated histidine that activates the attacking water (Figure 1B). This is supported by mutagenesis data that demonstrates H252 is key to the catalytic mechanism [39]. Both histidines are polarized by negatively charged side chains, E78 for H134 as before, and D212 for H252. A third acidic side chain, E39, coordinates the divalent cation from the previous mechanism. R9 (Q9 in human Dnase1) may stabilize the transition state. Along with the active site, this model assigned roles to key DNA binding residues that provide the energy relaxation needed for all these charged elements to occupy the confined active site via hydrogen bonding and Van der Waals interactions. The defined DNA-binding residues are E13, T14, R41, S43, N74, Y76, Y175, S206, T207, and Y211 [35]. Tyr76 is particularly important because it stacks against a nitrogenous base [35]. The key strengths of this model are the ability to explain newer mutagenesis results, a complete catalytic mechanism, and the support of a substrate-enzyme crystal structure. The principal weaknesses of this model are the unassigned roles of N170 and D168, and the lack of structural evidence for H252 activating the attacking water.
The Double-Divalent Cation catalytic model built upon the previous model. The updated model was based on mutagenesis studies performed on previously assigned active site residues, and corroborated the DNA binding residues [36,40,41]. The key finding was that E39 and D168 were both essential for activity (Figure 1C). D168 was assigned the role of metal binding, which developed the two-cation model active site. This model is characterized by both D168 and E39 binding cations. Between the two cations, the histidines perform acid-base catalysis exactly as described in the previous model (Figure 1C). Strengths of the double divalent cation model include assignment of roles to the most experimentally studied residues, support from molecular dynamics simulations, and explanation of both mutagenesis and structural data [35,36,40,41,42]. This model is supported by a similar proposed mechanism for the related EEP family member (17% sequence similarity), Human Apurinic/Apyrimidinic Endonuclease1 (hAPE1) at neutral pH [43]. This model also is supported by pKa studies of the active site histidines, via pH dependent imidazole rescue of Dnase1 activity for H134A and H252A mutants [40]. However, the Double-Divalent Cation model suffers from two key weaknesses. The role for the catalytically required N170 is unexplained, and there is no structural support for the two divalent cations in the Dnase1 active site.
The most recent model of Dnase1 catalysis is the Single Cation Carboxylic Acid catalysis model. This model was proposed based on a high-resolution structure solved during development of therapeutic Dnase1 for Cystic Fibrosis [34]. This model accounts for new structural evidence from the recombinant Dnase1 crystal structure (PDB:4AWN) showing a phosphate anion and Mg2+ cation together in the active site (Figure 1D) [34]. The Single Cation Carboxylic Acid catalysis model includes only one metal ion in the active site and assigns the role of the catalytic base to D168. Protonation of the leaving group is performed by a proton coordinated by E39 (Figure 1D). This model diverges from previous models to account for the orientation of the observed phosphate and Mg2+ in the active site. The strength of the Single Cation Carboxylic Acid catalysis model is its basis on a recent high-resolution substrate-cation structure. This model is consistent with a proposed hAPE1 mechanism [44,45]. Key weaknesses of this model are that it does not provide a biochemical mechanism for the protonation event and discounts previous experimental results highlighting the importance of the second histidine in the active site [39]. Furthermore, the proposed hAPE1 catalytic mechanism is controversial because the crystal structures support both a single cation-binding site and two cations binding in the active site, depending on pH [43]. Thus, there are currently two leading models for the mechanism by which Dnase1 cleaves DNA.
There remain many opportunities to better understand the mechanism of catalysis by Dnase1 family members. To date, the catalytic mechanism relies entirely on Dnase1 because Dnase1 is the only Dnase1 family member whose structure has been solved. Since the catalytic site is well conserved, it is likely that the mechanism will remain similar for all family members. Importantly, the key catalytic residues discussed here are identical across all family members [7]. However, despite this active site identity, Dnase1L2 is more active at an acidic pH. Additionally, Dnase1L3 has a unique C terminal domain not present in any other Dnase1 family or EEP superfamily member [4,18]. This domain enables digestion of dsDNA complexed with proteins and lipids [18,30], which could add to the catalytic mechanism. Finally, structural characterization of the Dnase1L3 inhibitors DR396, Pontacyl Violet, and Fmoc-D-cyclohexylalanine are still outstanding [46,47]. Thus, there are many opportunities to better understand this important enzyme family, especially with a broader focus beyond Dnase1.
3. The Pathophysiology of the Dnase1 Family
The absence of Dnase1 family members generally causes autoimmunity. Loss of the serum Dnases (Dnase1 and Dnase1L3) is associated with systemic autoimmunity, due to their role in clearing NETs (Dnase1 and Dnase1L3), degrading cell-associated DNA, and preventing anti-dsDNA antibody formation/urticarial vasculitis (Dnase1L3) (Figure 2A,B). Dnase1L1 deficiency is not yet definitively associated with any disease, but Dnase1L1 serves as the cellular receptor for pathogenic bacteria in the genus Ehrlichia (Figure 2C). Finally, absence of the skin-localized Dnase1L2 is associated with parakeratosis and psoriasis due to its role in clearing DNA from cells during cornification (Figure 2D).
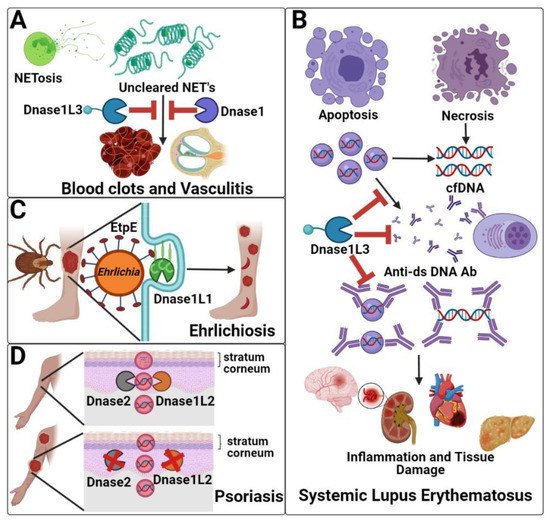
Figure 2. Dnase1 family members in disease. (A) NETosis releases chromatin into extracellular spaces. Dnase1 and Dnase1L3 absence may reduce NET clearance, which causes inflammation, resulting in blood clots and vasculitis. (B) Dnase1 and Dnase1L3 both process cfDNA. Dnase1L3 further digests DNA in microparticles, which prevents the anti-dsDNA autoantibody production and immune complex formation that leads to SLE. (C) After transmission by ticks, Ehrlichia spp. use EtpE to bind to the GPI anchor of Dnase1L1 for internalization and disease pathogenesis. (D) Dnase1L2 and Dnase2 deficiency is associated with psoriasis due to the failure to degrade the DNA within the nucleated cells in the stratum corneum during keratinocytes differentiation.
3.1. Clearance of Neutrophil Extracellular Traps (Dnase1/Dnase1L3)
One cause of inflammation and autoimmunity is the failure to clear the antimicrobial neutrophil extracellular traps (NETs). When NETs are not cleared and circulate at high levels, they damage the lungs, heart, and kidneys [48]. Reduced NET clearance is associated with vasculitis, psoriasis, SLE, pancreatitis and thrombosis [26,49,50,51]. Lack of NET clearance resulted in Factor XII (FXII) activation leading to pulmonary thrombo-inflammation [52]. NETs initiate and accrete thrombosis in blood vessels, which can develop into a severe complication during SARS-CoV2 infection [48,51]. This suggests complications during Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2) infection may be due to lack of clearance of NETs [48]. Thus, it is critical to remove NETs after pathogen destruction.
Dnase1 and Dnase1L3 are both critical to clearing NETs. In the absence of both Dnase1 and Dnase1L3, NET accumulation was reported to cause clots and vasculitis that leads to organ damage [49]. Either Dnase1 or Dnase1L3 was sufficient to prevent vasculitis [49,53] (Figure 2A). Targeting NETs and FXII activation during early onset of disease could potentially helpful prevent microvascular thrombosis [52]. Inhalation of Dnase1 may dissolve NETs in airways and improve symptoms during SARS-CoV2 infection [48,54]. Therefore, serum Dnases may be essential to return the body to homeostasis after NETosis.
3.2. Systemic Lupus Erythematosus (Dnase1/Dnase1L3)
Both serum Dnases are further implicated in systemic lupus erythematosus (SLE). SLE is characterized by chronic inflammation and production of autoreactive antibodies that target nuclear antigens, especially anti-double-stranded DNA (anti-dsDNA) antibodies [55,56,57]. Failure to degrade DNA promotes anti-dsDNA autoantibody production, immune complex formation, and deposition in multiple organs, leading to inflammation and organ failure (Figure 2B). Deficiency of Dnase1 alone on a mixed B6/129 background, which is predisposed to autoimmunity, causes lupus-like phenotypes in mice [58]. In contrast, Dnase1L3 deficiency causes lupus-like phenotypes in both mixed B6/129 mice and on a pure B6 background, indicating that Dnase1 cannot rescue Dnase1L3 loss [18]. Similarly, in humans, complete Dnase1L3 deficiency causes pediatric onset lupus with 100% penetrance [59], while reduction in Dnase1L3 activity and Dnase1 polymorphisms are associated with adult-onset, polygenic lupus [18,60,61,62,63]. Consistent with the failure of Dnase1 to ameliorate disease in the absence of Dnase1L3, clinical trials using Dnase1 to treat lupus nephritis patients failed [64]. However, exogenous Dnase1 degraded extracellular DNA, reduced proteinuria, and delayed anti-dsDNA antibody development and mortality in the lupus-prone NZB/NZW F1 mouse model [65]. Bovine Dnase1 showed limited efficacy in SLE patients without lupus nephritis [66]. These failures of Dnase1 suggest that Dnase1L3 performs unique functions in preventing lupus, potentially due to its C-terminal domain. The C-terminal domain enables Dnase1L3 to digest DNA complexed to lipids and proteins and nuclear DNA [18,30,60]. Neutralizing antibodies that target Dnase1L3 contribute to SLE pathogenesis in ~50% of sporadic cases [60]. Extracellular DNA degradation is likely key to Dnase1L3 preventing lupus onset. However, Dnase1L3 inhibition also prevents inflammasome formation [46]. The extent to which impairments in inflammasome contribute to SLE remains unknown. Similarly, if a threshold concentration of Dnase1L3 activity is needed to prevent disease, and how reduction in Dnase1 activity modifies other disease components, remain unknown.
3.3. Hypocomplementemic Urticarial Vasculitis (Dnase1L3)
Activity-reducing mutations in Dnase1L3 can also cause a second autoimmune disease independently of anti-dsDNA antibodies called Hypocomplementemic Urticarial Vasculitis Syndrome (HUVS). HUVS, or McDuffie syndrome, is characterized by recurrent urticaria along with dermal vasculitis, gastrointestinal tract manifestions, and polyserositis [67,68]. HUVS patients present with reduced C1q, C3, and C4 levels and the presence of anti-C1q; arthralgia or arthritis; uveitis or episcleritis; glomerulonephritis serum, but the absence of anti-dsDNA antibodies and anti-neutrophil cytoplasmic antibodies [68]. Instead, autoantibodies in HUVS are directed towards a collagen-like region on C1-q [68]. In rare cases, HUVS exhibits an immune-complex mediated glomerulonephritis with a membranoproliferative pattern of injury [69]. Due to the rarity of this disorder, little is known about the clinical manifestation, pathogenesis, treatment response, and outcome of such patients [68]. Moreover, 20% of HUVS patients exhibit shortness of breath, coughing, hemoptysis, pleural effusion [67,68]. Chronic obstructive pulmonary disease (COPD) is the most frequent cause of death among HUVS patients [67]. HUVS and SLE are strongly related because most HUVS patients progress to SLE later in life [62]. HUVS is associated with decreased Dnase1L3 activity [62,70]. For example, the homozygous frameshift mutation c.289_ 290delAC, in Dnase1L3 was present in three patients, while the homozygous exon skipping mutation c.320ⴙ4delAGTA was present in patients with unrelated families [62]. The c.320ⴙ4delAGTA skips exon 3, which codes for the first catalytic His in Dnase1L3. The mechanisms that predispose individuals with reduced Dnase1L3 activity to either HUVS, SLE or both remains unknown. The lack of animal models has limited our understanding of HUVS, and how it compares to SLE.
3.4. Ehrlichiosis (Dnase1L1)
While Dnase1L1 (Dnase X) deficiency is not yet definitively associated with any disease, Dnase1L1 is the cellular receptor for the causative agents of ehrlichiosis. Ehrlichiosis is caused by Gram-negative, obligate intracellular bacteria of the family Anaplasmataceae, such as Ehrlichia chaffeensis, Ehrlichia ewingii, and Ehrlichia muris eauclairensis [71,72]. These bacteria are transmitted to their human, canine or feline hosts primarily by ticks, such as the lone star tick (Amblyomma americanum) or blacklegged tick (Ixodes scapularis) [73]. Symptoms of ehrlichiosis include fever, headaches, rigor, myalgia, and malaise, which may take 4 weeks to develop [72]. This extended time to development of symptoms is a challenge for early detection of ehrlichiosis. Symptoms caused by ehrlichiosis, including leukopenia or anemia, can also become severe, especially when left untreated or in elderly and immunocompromised populations [74]. Thus, early detection of ehrlichiosis is crucial so that patients can be quickly treated with antibiotics, such as doxycycline [72].
Dnase1L1 has a key role in the early stages of ehrlichiosis because it serves as the surface receptor engaged by Ehrlichia spp. Ehrlichia spp. infect host phagocytes, such as neutrophils, by targeting the glycosylphosphatidylinositol (GPI) anchor in Dnase1L1 [75,76,77,78] (Figure 2C). Internalization is facilitated by the Ehrlichia protein EtpE binding to Dnase1L1 [78]. Once phagocytosed, E. chaffeensis prevents phagolysosomal fusion [74]. Ehrlichia spp. replicate inside cytoplasmic vacuoles and form dense microcolonies called morulae [72,74]. Ehrlichia spp. use Dnase1L1 to block reactive oxygen species generation [77]. This is done by preventing the production of the NADPH Oxidase 2 complex that generates superoxide (O2−) from oxygen [77]. Thus, Ehrlichia spp. promote disease by hijacking Dnase1L1.
Interestingly, the involvement of Dnase1L1 in ehrlichiosis may implicate Dnase1L1 in diabetes. Diabetic retinopathy could be caused in part by ehrlichiosis [79]. High levels of Dnase1L1 expression are further related to diabetes [80,81]. Patients with type 2 diabetes had higher pancreatic Dnase1L1 expression compared to patients without type 2 diabetes [81]. Quantitative Trait Loci mapping also revealed a correlation between Dnase1L1 and diabetes [80]. Thus, there is an opportunity to determine what role, if any, Dnase1L1 plays in diabetes, and if Dnase1L1 connects ehrlichiosis to diabetes.
3.5. Psoriasis (Dnase1L2)
Deficiency in Dnase1L2 is associated with psoriasis and parakeratosis. Parakeratosis is the persistence of nucleated cells in the stratum corneum [82]. Parakeratosis is directly linked to psoriatic lesions. Psoriasis is an autoimmune disease characterized by chronic skin inflammation. There are two distinct types of psoriasis: chronic plaque psoriasis and generalized pustular psoriasis. Chronic plaque psoriasis stems from the adaptive immune system and is characterized by erythematous scaly plaques. One major genetic risk factor is the Cw*0602 MHC class I allele [83]. This MHC allele stimulates pro-inflammatory cytokine production, which contribute to chronic plaque psoriatic inflammation [83]. In contrast, generalized pustular psoriasis stems from the innate immune system and is characterized by red pustules [83]. Dnase1L2 deficiency causes parakeratosis [84]. Activity-reducing Dnase1L2 SNPs may be associated with psoriasis [85], and psoriatic lesions have reduced Dnase1L2 [17]. In the absence of Dnase1L2, DNA is not digested during keratinocyte differentiation, so corneocytes retain their nuclear DNA throughout the stratum corneum [84]. Dnase1L2 primarily contributes to terminal keratinocyte differentiation, which is why the enzyme is 16 times more concentrated in the skin than in any other organ [17] (Figure 2D). When comparing Dnase1L2−/− mice to wild type mice, Dnase1L2−/− mice had 400× more nuclear DNA in their hair and 2000× more nuclear DNA in nails compared to wild type mice [84]. The intracellular Dnase Trex2 cooperates with Dnase1L2 to degrade DNA in the oral epithelium [86]. Similarly, Dnase2 is also active in the skin [87]. However, sufficient Dnase2 levels were present in psoriatic scales, but Dnase1L2 was reduced [87]. Overall, this suggests that Dnase1L2 is a key Dnase to prevent psoriasis, but that psoriasis requires multiple hits [87]. Dnase1L2 acts optimally at an acidic pH, consistent with maximal activity during or just after cornification [17]. To improve our understanding of the clinical implications of Dnase1L2 deficiency, human skin models are needed to complement mouse models. Depletion of Dnase1L2 and/or Dnase2 in human skin will provide new insights into their relative contribution to parakeratosis and psoriasis in humans. Discerning the role of Dnase1L2 in psoriasis may provide new therapeutic targets to treat psoriasis.
4. Clinical and Experimental Applications of Dnases
4.1. Experimental Applications of Dnase1
The importance of the catalytic mechanism is underscored by the widespread use of Dnase1. Dnase1 is a potent experimental research tool and clinical treatment. In the lab, Dnase1 is used to remove unwanted nucleic acids from experiments. For example, Dnase1 is widely used to treat RNA preparations to degrade any remaining genomic DNA. This is important because contaminating DNA can cause false positives in RT-PCR. Dnase1 is central to DNase footprinting assays, because the inability of Dnase1 to degrade protein-bound DNA reveals the DNA-binding site of proteins [88]. Finally, Dnase1 is used as a control in DNA degradation and activity assays. Thus, Dnase1 is a potent research tool.
4.2. Clinical Applications of Dnase1
Dnase1 is used in the clinic, most notably as dornase alfa (trade name Pulmozyme) to treat Cystic Fibrosis (CF). During CF, extracellular DNA accumulates in the lungs, which increases mucosal viscosity and impedes mucociliary clearance and pulmonary function [89]. Recombinant Dnase1 is administered in CF patients to improve mucociliary clearance and pulmonary function [89,90,91]. Dnase1L2 is under development as a next-generation treatment for CF [92]. Beyond CF, Dnase1 has been tested in other pulmonary diseases, including asthma, obstructive pulmonary disease, chronic pulmonary diseases, and hemorrhagic shock [93,94,95]. The efficacy of Dnase1 in these other diseases has been mixed [95], though mouse models have shown more promising efficacy. Dnase1 was protective in a mouse model of asthma [94]. In a hemorrhagic shock mouse model, Dnase1 treatment attenuated tissue damage and systemic/lung inflammation, improving survival [93]. However, the efficacy of Dnase1 in asthma, especially in pediatric patients, is controversial [95], suggesting more work is needed on this potential therapy.
Dnases have been explored in mouse models for efficacy against diseases caused by defects in NET clearance. Dnase1 clears NETs in vivo [23]. Intravenous administration of Dnase1 improved mouse survival during Escherichia coli-induced sepsis due to NET cleavage [96]. Dnase1 treatment also protected mice in a model of deep vein thrombosis [97]. During liver injury, release of DNA exacerbates injury. Dnase1 treatment reduced symptoms in thioacetamide-induced hepatorenal injury, while Dnase1L3 may contribute to protection during acetaminophen-induced hepatic injury [98,99]. Overall, Dnases have seen success in pre-clinical mouse models to reduce symptoms from pathogenic DNA. However, it remains to be seen if these successes can be replicated in humans.
Finally, a case report suggested that a patient with severe end stage Alzheimer’s disease had significant symptomatic improvement upon Dnase1 treatment [100]. Overall, Dnase1 family members have a wide application in the clinic to treat disease, especially pulmonary diseases.
4.3. Dnase1 Family Members and Cell-Free DNA as Potential Biomarkers
Along with being used to treat disease, both Dnase1 family members and their cleaved DNA products are useful as biomarkers for disease. Changes in Dnase levels/activity are observed in autoimmunity, asthma, myocardial infarction, and cancer. In the cases of asthma, myocardial infarction, and cancer, it is unclear if Dnase changes are responses to disease, cause disease onset, or impact disease at all. Defects in Dnase1 family activity cause autoimmunity [59]. Specifically, the activity-reducing Dnase1L3 mutation R206C is associated with rheumatoid arthritis, scleroderma, HUVS, and SLE [62,101,102,103]. Decreased Dnase1L3 activity is associated with SLE, dermatomyositis, and polymyositis, but not rheumatoid arthritis, despite association of the gene with rheumatoid arthritis [18,59,60,61,104,105]. Dnase1 polymorphisms have also been reported in SLE patients [63]. Inflammatory bowel disease patients show reduced Dnase1L3 levels, but not to the extent of SLE patients [106]. Similarly, Dnase1L3 is downregulated in pneumonia caused by Mycoplasma pneumoniae [107]. This contrasts with asthma, where elevated sputum Dnase1L3 has been validated as one of six biomarkers in a randomized controlled trial to detect and discriminate between forms of asthma [108]. Dnase1L3 intronic variants are associated with risk and exacerbation of asthma, especially in patients of African descent [109]. Serum Dnase1 is also elevated during myocardial infarction after the onset of chest pain [110]. The Dnase1*2 allele, which includes a Q222R mutation, is associated with myocardial infarction in Japanese patients [111]. In contrast, Dnase1L3 is usually decreased in cancer. Dnase1L3 levels are reduced in breast cancer, cholangiocarcinoma, lung adenocarcinoma, hepatocellular carcinoma, and clear cell renal cell carcinoma [112,113,114]. Low levels of Dnase1L3 are associated with a worse prognosis in breast, kidney, and stomach cancer, hepatocellular carcinoma, lung adenocarcinoma, and sarcoma, while higher levels were associated with increased overall survival in hepatocellular carcinoma [112,114]. However, hepatitis B-related hepatic cancer shows increased levels of Dnase1L3 [115]. The Dnase1*2 allele may be a good predictor of gastric and colorectal carcinoma development, though no association was found with other cancers [96]. Overall, Dnase1 family members, especially Dnase1 and Dnase1L3, are potential biomarkers for autoimmunity, asthma, myocardial infarction, and cancer.
In addition to Dnases themselves as biomarkers for disease, the circulating serum cell-free DNA (cfDNA) edited by Dnases can serve as a biomarker. Cell-free DNA is unencapsulated DNA within the blood, produced by necrosis and programmed cell death. Dnase1 and Dnase1L3 both digest cfDNA with characteristic traits. Dnase1 tends to reduce the amount of single-stranded DNA on the end of dsDNA fragments, or “jaggedness”, while Dnase1L3 increases jaggedness [116]. Dnase1L3 also creates “CC” end motifs on DNA fragments [117]. Thus, serum Dnases process circulating cfDNA with characteristic signatures.
New technologies have enabled the testing and tracking of cfDNA. Fluorometric assays, real-time PCR, and sequencing allow identification and analysis of cfDNA in plasma. Analysis of cfDNA in plasma has been branded as a “liquid biopsy,” because cfDNA now serves as a biomarker for a wide range of conditions and diseases, including prenatal testing, inflammatory bowel disease, autoimmunity, and cancer [117,118,119,120,121,122]. High levels of serum cfDNA in SLE patients were first observed in 1966 [55]. Cell-free DNA as a biomarker for metastatic cancer was proposed in 1977 [123]. Today, cfDNA is used for diagnosis, monitoring, and determining prognosis for cancer patients [120,124]. Sequencing of tumor cfDNA enables identification of tumor genetics and drug resistance [124]. One challenge with cfDNA as a biomarker is that cfDNA levels vary widely amongst cancer patients [125]. No standardized baseline for cancer testing has yet been developed due this variability and due to the multiple possible sources of cfDNA [125]. Overall, cfDNA represents a promising biomarker, which can be modified by Dnase1 activity.
This entry is offline, you can click here to edit this entry!