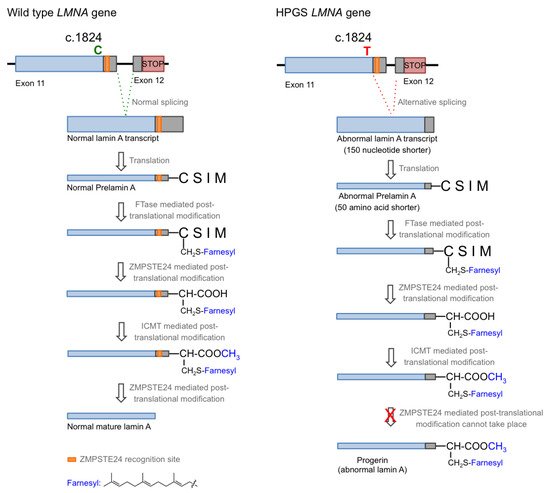
In the normal post-translational prelamin A maturation process, the cytosolic enzyme FTase adds a 15-carbon farnesyl lipid to prelamin A that is eventually removed after the four-step translational processing suffered by this protein, schematized in
Figure 1. However, the presence of the mutation prevents the last hydrolytic cleavage (
Figure 1) yielding permanently farnesylated progerin. The presence of this group increases the hydrophobicity of the protein, thus promoting strong interactions with the nuclear membrane, where its abnormal accumulation severely affects the normal nuclear functions. Early studies described that blocking or preventing farnesylation improved nuclear abnormalities in mouse and human progeroid fibroblasts and reduced nuclear blebbing and the number of misshapen nuclei [
11,
39,
40]. Furthermore, in vivo treatment with an FTase inhibitor (FTI) ameliorated the progeria phenotype in the Zmpste24-deficient mouse model of progeria [
41]. Accordingly, inhibition of FTase was one of the first therapeutic strategies suggested for ameliorating the severity of the disease [
42] and the only one that, up to date, has allowed the first approved drug for the specific treatment of progeria to be developed. FTIs were initially developed as anticancer compounds, aimed at preventing the permanent Ras activation characteristic of the frequent and deadly Ras-driven tumors [
43]. The rational of this therapeutic potential was based on the fact that in the absence of farnesylation, Ras would be unable to attach to the cell membrane [
44]. Hence, in the presence of an FTI, Ras should be inactive. Although the lack of efficacy of FTIs in phase III clinical trials stopped their advance into the clinic, the similarity between the post-translational processing of progerin and Ras (both belong to the CAAX family of proteins) set up the bases for the repurposing of FTIs for treating progeria by reducing the levels of farnesylated progerin. Among the different FTIs showing high efficacy in the inhibition of FTase in vivo, lonafarnib (
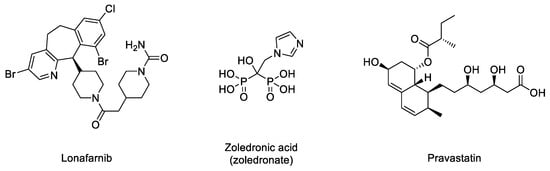
Figure 3) soon stood out as the most promising candidate to start a single-arm phase II clinical trial in 2007 (NCT00425607) [
45]. This trial showed that lonafarnib was well tolerated, and it also showed the capacity of this compound to improve some of the symptoms of the disease, such as rate of weight gain, arterial pulse wave velocity, carotid artery echodensity, skeletal rigidity, and sensorineural hearing. Importantly, it also decreased mortality rate (3.7% vs. 33.3% after a median of 2.2 years of follow-up in individuals receiving lonafarnib monotherapy compared with no treatment) [
46]. Although lonafarnib does not correct all the alterations of the disease, such as lipodystrophy, skin features, alopecia, and joint contractures, the fact that it is the only available drug for treating this lethal pathology and that it has a positive impact in ameliorating specific features of HGPS, has led to its recent approval by the FDA under the name of Zokinvy
TM [
47].
There may be various reasons for lonafarnib’s limited improvement. One of the first hypotheses suggested that in the absence of FTase activity, alternative prenylation by the enzyme geranyl geranyltransferase (GGTase) could take place, as this mechanism had been already characterized in the limited Ras inactivation observed in vivo after treatment with FTIs. Hence, it was suggested that the concomitant inhibition of both enzymes could increase the efficacy of the treatment. The phenotypic improvement observed in an in vivo model of progeria after the combination of the statin pravastatin and the aminobisphosphonate zoledronate seemed to confirm this idea [
48] and was the basis for starting a triple therapy clinical trial in which lonafarnib, pravastatin, and zoledronic acid were administered to progeria patients (clinical trials NCT00879034 and NCT00916747). Although no participants withdrew because of side effects, no significant improvements other than increased bone mineral density were observed compared to lonafarnib monotherapy. Hence, as the triple therapy did not convey any obvious advantage over monotherapy, it was discontinued, and the clinical trial was prolonged but only with lonafarnib administration [
49]. In addition, it is possible that other targets of FTase, apart from progerin, are affected by lonafarnib, eliciting adverse effects in progeroid patients. In line with this concern, since lonafarnib was originally developed for the treatment of Ras-dependent tumors [
44], it is antiproliferative, a feature that can potentially limit its positive effects on progeroid cells, in which pro-proliferative effects are needed. In this regard, it has been reported that after long-term treatments, lonafarnib induced cell death and formation of donut-shaped nuclei [
50,
51]. These negative effects should be taken into consideration, especially when analyzing the effects of combining lonafarnib with other potential anti-progeroid therapies [
51].