Statins are the gold standard treatment in patients with familial hypercholesterolemia due to their effectiveness reducing circulating cholesterol levels and their low cost. However, increasing evidence shows a clear relation between long term statin treatment and new-onset type 2 diabetes mellitus (T2DM) development. However, the mechanisms by which statin treatment induces T2DM are not fully understood. Recent evidences suggest that statins could increase circulating glucose levels inducing insulin resistance in peripheral tissues and modulating insulin sensitivity and secretion in beta-cells. It has been described that calcium signaling, a key process that allows insulin-containing vesicle mobilization is impaired in the beta-cells under statin treatment, the expression and mobilization of glucose transporters are also reduced and insulin signaling pathway is down regulated after statin treatment. All together contribute to glucose and insulin homeostasis deregulation and favors T2DM development.
- β-cells, diabetes, statin
Insulin secretion from pancreatic β-cells is initiated by glucose-induced Ca2+ entry controlled by voltage-gated Ca2+ channels [1]. Therefore, maintenance of intracellular Ca2+ homeostasis is tightly regulated in order to ensure proper insulin secretion and maintain the integrity of the β-cell physiology [2]. Briefly, glucose uptake activates glycolysis in β-cell thus elevating the [ATP]/[ADP]i ratio [3]. This acts as a signal that closes KATP channels and depolarizes the plasma membrane, with subsequent activation of voltage-dependent Ca2+ channels, entry of extracellular Ca2+ and finally insulin exocytosis [4]. ATP sensitivity of the KATP channels is modulated by several effectors including PIP2 and acyl CoAs [5][6]. Conversely, a decrease in the metabolic signal causes reopening of KATP channels and suppresses the electrical trigger for insulin secretion, thereby providing feedback regulation of insulin secretion [7]. In addition, ATP and ADP can act as autocrine activators of β-cell purinergic receptors because they are also within insulin exocytosis granules [8]. Indeed, inhibition of both P2X and P2Y purinergic receptors causes a reduction in glucose-induced insulin secretion [9][10][11][12].
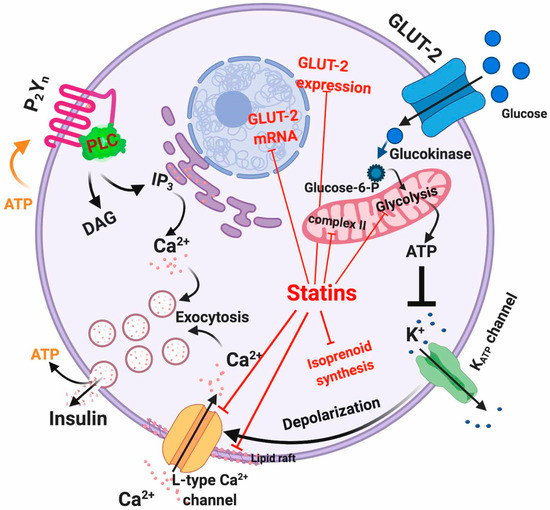
Figure 1. Intracellular actions of statins in β-cells. Red lines indicate the mechanisms affected by statins.
To date, the relationship between statin-mediated inhibition of cholesterol synthesis and impaired L-type Ca2+ channel activity remains unclear. However, in vitro studies have indicated that simvastatin can directly inhibit L-type Ca2+ channels in rat pancreatic islet β-cells [13]. Specifically, because simvastatin was found to immediately inhibit channel activity, it has been suggested that there is a direct interaction between simvastatin and the channel. In contrast, pravastatin lacks L-type Ca2+ channels inhibition, possibly because of its lipophilicity [13]. Alternatively, other authors have suggested that the long-term cholesterol reduction caused by statins can lead to incorrect sorting of membrane lipid-raft bound proteins or conformational changes of the Ca2+ channel subunits [14]. More recently, it has been suggested that statins can reduce the membrane potential by inhibiting mitochondrial complex II activity, which causes oxidative stress [15]. These off-target effects of statins have been very recently corroborated by Curry et al. [16] in experiments showing that simvastatin impairs β-cell function by at least two mechanisms: (1) via direct inhibition of KATP channels in a mitochondria-independent manner and (2) via interference with mitochondrial respiration, thus decreasing cytosolic ATP levels and inhibiting metabolic upregulation of L-type Ca2+ channels [16].
As described before, insulin is secreted by β-cells in response to glucose uptake through GLUT receptors (primarily GLUT-1 to 4), with GLUT-2 being the predominant isoform in β-cells [17][18]. GLUT-2 represents a high-affinity and low-capacity glucose transporter [19]. It has been shown that treatment of β-cells with atorvastatin and pravastatin inhibited GLUT-2 expression in a concentration-dependent manner [20]. However, rosuvastatin and pitavastatin showed a slight increase in GLUT-2 expression [20]. In addition to this, it has also been observed in mouse pancreatic β-cell line MIN6 cells that simvastatin treatment diminishes GLUT-2 mRNA and protein expression via a dose-dependent reduction of ATP production [21]. Another mechanism through which statins may interfere with glucose metabolism is the statin-mediated LDLR upregulation that increases cholesterol uptake in the β-cell leading to reduced mRNA and protein expression of GLUT-2, consequently limiting glucose uptake [22][23].
The direct inhibition of the mevalonate pathway by statins reduces the intracellular concentration of isoprenoids, the final products of the pathway. Isoprenoids are essential for G protein posttranslational modification, which is important for insulin granule exocytosis [24]. Interestingly, it has been shown that the glucose-induced insulin secretion by lovastatin in normal rat islets is reduced by co-incubation with mevalonate [25].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21134725
References
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P.; Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. . Diabetes 2008, 57, 1618–1628, .
- Jahanshahi, P.; Wu, R.; Carter, J.D.; Nunemaker, C.S.; Evidence of diminished glucose stimulation and endoplasmic reticulum function in nonoscillatory pancreatic islets. . Endocrinology 2009, 150, 607–615, .
- Rorsman, P.; Eliasson, L.; Renstrom, E.; Gromada, J.; Barg, S.; Gopel, S.; The Cell Physiology of Biphasic Insulin Secretion. . News Physiol. Sci. 2000, 15, 72–77, .
- MacDonald, P.E.; Wheeler, M.B.; Voltage-dependent K(+) channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. . Diabetologia 2003, 46, 1046–1062, .
- Fridlyand, L.E.; Tamarina, N.; Philipson, L.H.; Bursting and calcium oscillations in pancreatic beta-cells: Specific pacemakers for specific mechanisms. . Am. J. Physiol. Endocrinol. Metab. 2010, 299, E517–E532, .
- Gilon, P.; Shepherd, R.M.; Henquin, J.C.; Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. . J. Biol. Chem. 1993, 268, 22265–22268, .
- Ashcroft, F.M.; Rorsman, P.; K(ATP) channels and islet hormone secretion: New insights and controversies. . Nat. Rev. Endocrinol. 2013, 9, 660–669, .
- Bertrand, G.; Chapal, J.; Loubatieres-Mariani, M.M.; Potentiating synergism between adenosine diphosphate or triphosphate and acetylcholine on insulin secretion. . Am. J. Physiol. 1986, 251, E416–E421, .
- Geisler, J.C.; Corbin, K.L.; Li, Q.; Feranchak, A.P.; Nunemaker, C.S.; Li, C.; Vesicular nucleotide transporter-mediated ATP release regulates insulin secretion. . Endocrinology 2013,, 154, 675–684, .
- Jacques-Silva, M.C.; Correa-Medina, M.; Cabrera, O.; Rodriguez-Diaz, R.; Makeeva, N.; Fachado, A.; Diez, J.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; et al. Jacques-Silva, M.C.; Correa-Medina, M.; Cabrera, O.; Rodriguez-Diaz, R.; Makeeva, N.; Fachado, A.; Diez, J.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; et al. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc. Natl. Acad. Sci. USA 2010, 107, 6465–6470. Proc. Natl. Acad. Sci. USA 2010, 107, 6465–6470, .
- Khan, S.; Yan-Do, R.; Duong, E.; Wu, X.; Bautista, A.; Cheley, S.; MacDonald, P.E.; Braun, M.; Autocrine activation of P2Y1 receptors couples Ca (2+) influx to Ca (2+) release in human pancreatic beta cells. Diabetologia 2014, 57, 2535–2545.. Diabetologia 2014 , 57, 2535–2545, .
- Wuttke, A.; Idevall-Hagren, O.; Tengholm, A.; P2Y(1) receptor-dependent diacylglycerol signaling microdomains in beta cells promote insulin secretion. . FASEB J. 2013, 27, 1610–1620, .
- Yada, T.; Nakata, M.; Shiraishi, T.; Kakei, M.; Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. . Br. J. Pharm. 1999 , 126, 1205–1213, .
- Xia, F.; Xie, L.; Mihic, A.; Gao, X.; Chen, Y.; Gaisano, H.Y.; Tsushima, R.G.; Inhibition of cholesterol biosynthesis impairs insulin secretion and voltage-gated calcium channel function in pancreatic beta-cells. . Endocrinology 2008, 149, 5136–5145, .
- Sadighara, M.; Amirsheardost, Z.; Minaiyan, M.; Hajhashemi, V.; Naserzadeh, P.; Salimi, A.; Seydi, E.; Pourahmad, J.; Toxicity of Atorvastatin on Pancreas Mitochondria: A Justification for Increased Risk of Diabetes Mellitus. . Basic Clin. Pharm. Toxicol. 2017 , 120, 131–137, .
- Curry, L.; Almukhtar, H.; Alahmed, J.; Roberts, R.; Smith, P.A.; Simvastatin Inhibits L-Type Ca2+-Channel Activity Through Impairment of Mitochondrial Function. . Toxicol. Sci. 2019, 169, 543–552, .
- Gould, G.W.; Holman, G.D.; The glucose transporter family: Structure, function and tissue-specific expression. . Biochem. J. 1993, 295 Pt 2, 329–341, .
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F.; Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. . Cell 1988, 55, 281–290, .
- Baudry, A.; Leroux, L.; Jackerott, M.; Joshi, R.L.; Genetic manipulation of insulin signaling, action and secretion in mice. Insights into glucose homeostasis and pathogenesis of type 2 diabetes. . EMBO Rep. 2002, 3, 323–328, .
- Zhao, W.; Zhao, S.P.; Different effects of statins on induction of diabetes mellitus: An experimental study. . Drug Des. Dev. Ther. 2015, 9 , 6211–6223, .
- Zhou, J.; Li, W.; Xie, Q.; Hou, Y.; Zhan, S.; Yang, X.; Xu, X.; Cai, J.; Huang, Z.; Effects of simvastatin on glucose metabolism in mouse MIN6 cells. . J. Diabetes Res. 2014, 2014, 376570, .
- Kruit, J.K.; Brunham, L.R.; Verchere, C.B.; Hayden, M.R.; HDL and LDL cholesterol significantly influence beta-cell function in type 2 diabetes mellitus. . Curr. Opin. Lipidol. 2010, 21 , 178–185, .
- Roehrich, M.E.; Mooser, V.; Lenain, V.; Herz, J.; Nimpf, J.; Azhar, S.; Bideau, M.; Capponi, A.; Nicod, P.; Haefliger, J.A.; et al. Insulin-secreting beta-cell dysfunction induced by human lipoproteins. . J. Biol. Chem. 2003 , 278, 18368–18375, .
- Betteridge, D.J.; Carmena, R.; The diabetogenic action of statins-mechanisms and clinical implications. . Nat. Rev. Endocrinol. 2016, 12, 99–110, .
- Metz, S.A.; Rabaglia, M.E.; Stock, J.B.; Kowluru, A.; Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. . Biochem. J. 1993, 295 Pt 1, 31–40, .