Neurodegeneration in multiple sclerosis is believed to underlie disease progression and permanent disability. Many mechanisms of neurodegeneration in MS have been proposed, such as mitochondrial dysfunction, oxidative stress, neuroinflammation, and most recently RNA-binding protein dysfunction. Studying RNA-binding protein dysfunction addresses a gap in our understanding of the pathogenesis of MS, which may allow for novel therapies to be generated to attenuate neurodegeneration before irreversible central nervous system damage occurs.
- Multiple Sclerosis
- Neurodegeneration
- RNA binding protein dysfunction
- Heterogenous nuclear ribonucleoprotein A1
Introduction
Multiple sclerosis (MS) is a demyelinating, autoimmune, and neurodegenerative disease of the central nervous system (CNS) that results in a variety of physical and cognitive deficits. The etiology of MS is largely unknown; however, it is believed that an environmental factor combined with genetic predisposition triggers an autoimmune response to CNS antigens.The majority of MS patients initially present with a relapsing-remitting disease course, where relapses of clinical symptoms are followed by a period of remission in which symptoms partially or completely recover. The clinical manifestations of relapses are considered to be a result of acute inflammation and demyelination [1]. Over time, a significant proportion of relapsing-remitting MS patients will develop a progressive form of the disease (secondary-progressive MS) involving continuous worsening of neurological symptoms. A small percentage of patients present initially with primary-progressive MS where disease continuously progresses without remission. Ongoing neurodegeneration is characteristic of progressive disease and is believed to be responsible for the permanent, irreversible damage in MS patients [2-4]. The clinical significance of neurodegeneration is important because it has a stronger correlation with clinical disability than demyelination [5-8].
Most of the treatments for MS are immunotherapies that act to regulate an overactive immune system. These drugs have been proven to be effective in offering a better quality of life for the majority of patients, specifically those with relapsing-remitting MS [9,10]. The reason why these therapies might not significantly attenuate progressive disease is because they fail to disrupt the neurodegenerative process [11]. The cause of neurodegeneration in MS is predominately unknown, but is likely complex and multifactorial. Many mechanisms have been proposed and researched in depth, such as the increased energy requirements, sodium and calcium channel redistribution, and cellular stress that results from the demyelination of axons [12-14] as well as mitochondrial dysfunction, oxidative stress, and neuroinflammation. Recent data suggests that dysfunctional RNA-binding proteins (RBPs) may contribute to the pathogenesis of neurodegeneration in MS and its models, which is the focus of this review,
Dysfunctional RNA binding proteins
RBPs are highly conserved proteins whose expression is tightly regulated and necessary for cell survival. They play important roles in the regulation of gene expression as well as mRNA stability, splicing, and transport. Under homeostatic conditions, many RBPs localize to the nucleus where they continuously shuttle back and forth to the cytoplasm; a process mediated by nuclear export and nuclear localization signals within the RBP. RBPs contain functional regions that have the ability to bind RNA as well as other proteins creating higher order structures. Under homeostatic circumstances, RNAs and RBPs can be concentrate into membraneless structures such as stress granules (SGs), which generally contain a denser core and a more dynamic shell that interacts with the surrounding cytoplasm [15]. Cytoplasmic SGs are dynamic, membraneless granules that harbor translationally repressed mRNAs and proteins, including RBPs, as a mechanism of protection. SG formation alters patterns of local RNA translation facilitating the stress response [16]. Under physiological conditions, once the acute stressor is removed, SGs disassemble and translation resumes. In contrast, under pathologic conditions, SGs may persist leading to decreased cell survival and the induction of proapoptotic pathways [17–20]. Abnormally stable SGs may serve as a platform for formation of larger protein aggregates, and further interactions between RNA and RBPs within SGs may change their dynamics, leading to a lack of disassembly [21]. The prolonged sequestration of important RBPs and mRNAs needed for cell survival combined with translational impairment might also be a mechanism of cell death [22]. SGs have been tightly linked to neurodegeneration in human neurologic diseases as well as their models [23-25].
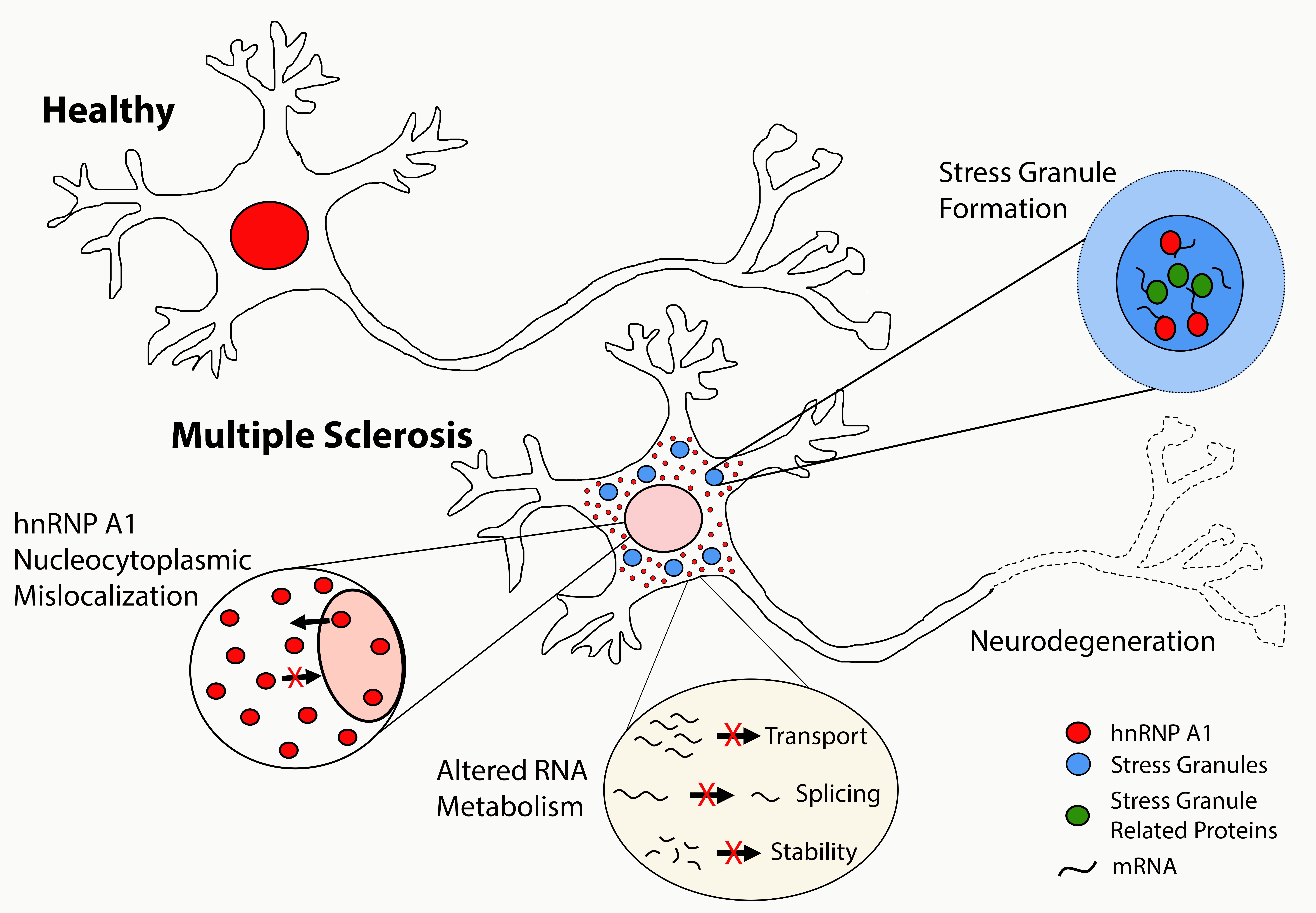
Figure: Dysfunctional RBPs result in a triad of molecular changes, including mislocalization of RBPs from its homeostatic nuclear location to the cytoplasm, co-localization in cytoplasmic stress granules and altered RNA metabolism.
Dysfunctional RNA binding proteins in MS
Several factors have been found to disrupt RBP function, including mutations within RBPs, osmotic stress, oxidative stress, heat stress, and proinflammatory cytokines [25-27]. For example, in vitro experiments demonstrated that somatic mutations within the RBP heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) identified from MS patients contribute to RBP dysfunction [28]. These experiments showed that transfection of plasmids containing hnRNP A1 mutants in neuron-like cells resulted in hnRNP A1 mislocalization, colocalization into cytoplasmic SGs, and apoptosis, compared to cells transfected with plasmids containing wild type hnRNP A1. Considering inherited mutations within RBPs and subsequent RBP dysfunction were shown to underlie ALS and FTLD pathology [24,29-33], these experiments suggest similar mechanisms may contribute to the pathogenesis of MS.
There is substantial evidence demonstrating RBP dysfunction in neurons in MS and its models suggesting this may be an underlying mechanism of neurodegeneration. For example, hnRNP A1 mislocalization and SG formation, two of the pathological hallmarks of dysfunctional RBPs, were initially shown in neurons from a single MS case [26]. Recently, these findings were expanded to include 12 additional MS cases and six control cases, and demonstrated increased nucleocytoplasmic mislocalization of hnRNP A1 and the RBP Tar DNA Binding Protein 43 (TDP-43) in neurons from MS patients compared to controls [34]. Increased nucleocytoplasmic localization of RBPs has been shown to be toxic to neurons [35,36]. In a separate study, altered RBP biology was identified in oligodendrocytes and neurons of MS cases. These data found altered expression of the RBPs TDP-43 and polypyrimidine tract-binding protein 1 and 2 (PTB1 / PTB2) in cortical demyelinated lesions [37]. Considering the importance of TDP-43 and PTB1/2 in oligodendrocyte viability and neuronal differentiation, respectively, researchers hypothesized RBP dysfunction might contribute to cortical lesion damage and neurodegeneration in MS [37].
Additional experiments from the experimental autoimmune encephalomyelitis (EAE) animal models of MS have provided evidence of dysfunctional RBPs contributing to neurodegeneration. EAE mice showed increased hnRNP A1 and TDP-43 mislocalization and SG formation in neurons of the spinal cord, the former of which correlated with neurodegeneration [23,25]. Additionally, hnRNP A1 mislocalization was found to correlate with disease severity as well as with IFNγ-producing T cell infiltrates in the spinal cord of mice with EAE [25]. Furthermore, there was a significant negative correlation between the number of neurons in the spinal cord and hnRNP A1 mislocalization, indicating that there are fewer neurons in areas with increased hnRNP A1 mislocalization [25]. These experiments suggest a relationship between neurodegeneration, inflammation, and RBP dysfunction.
In addition to cytokine-induced mechanisms of RBP dysfunction, our lab has implicated an antibody response to hnRNP A1 as a mechanism of neurodegeneration in MS [23,38-43]. MS patients develop IgG specific for hnRNP A1, specifically to the M9 region of hnRNP A1, that is responsible for its transport into and out of the nucleus [44]. Previously published data demonstrated that peripheral injections of anti-hnRNP A1 antibodies which overlap with the immunodominant epitope of MS IgG, into mice with EAE resulted in worsening of disease, increased neurodegeneration, and a change in phenotype from flaccid to spastic hind limbs [45]. Spasticity of limbs is a common symptom of MS [46,47]. Immunohistochemical localization of anti-hnRNP A1 antibodies injected into mice with EAE showed antibody deposition within and surrounding spinal cord neurons. Additionally, we found that peripheral injection of anti-hnRNP A1 antibodies also exacerbated hnRNP A1 dysfunction [23,45]. Anti-hnRNP A1 antibodies were localized to areas of the spinal cord with increased neuronal hnRNP A1 nucleocytoplasmic mislocalization, SG formation, and neuronal loss compared to controls. This study demonstrated that, in addition to the neuroinflammatory response, antibodies to hnRNP A1, an intraneuronal target, augments RBP dysfunction and neurodegeneration in an animal model of MS.
Conclusions
Research suggests that dysfunctional RBPs are a pathologic hallmark of MS and its models and may contribute to neurodegeneration in mechanisms similar to other neurologic diseases. Further research involving dysfunctional RBPs is necessary to better understand the cellular pathways effected, so that precise therapeutic interventions can be created to prevent, attenuate, or reverse RBP dysfunction and in turn, alter the natural history of neurodegeneration and disease progression in MS.
Cole D. Libner, Hannah E. Salapa, Michael C. Levin.
Office of the Saskatchewan MS Clinical Research Chair, University of Saskatchewan
https://research-groups.usask.ca/skms-office/
This entry is adapted from the peer-reviewed paper 10.3390/ijms21134571
References
- Luessi, F.; Siffrin, V.; Zipp, F. Neurodegeneration in multiple sclerosis: Novel treatment strategies. Expert Rev. Neurother. 2012, 12, 1061–1077. [Google Scholar] [CrossRef]
- Levin, M.C.; Douglas, J.N.; Meyers, L.; Lee, S.; Shin, Y.; Gardner, L.A. Neurodegeneration in multiple sclerosis involves multiple pathogenic mechanisms. Degener Neurol Neuromuscul Dis. 2014, 4, 49–63. [Google Scholar] [CrossRef]
- Trapp, B.D.; Ransohoff, R.M.; Fisher, E.; Rudick, R.A. Neurodegeneration in multiple sclerosis: Relationship to neurological disability. Neuroscience 1999, 5, 48–57. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Anderson, V.M.; Fisniku, L.K.; Altmann, D.R.; Thompson, A.J.; Miller, D.H. MRI measures show significant cerebellar gray matter volume loss in multiple sclerosis and are associated with cerebellar dysfunction. Mult. Scler. J. 2009, 15, 811–817. [Google Scholar] [CrossRef]
- Fisniku, L.K.; Chard, D.T.; Jackson, J.S.; Anderson, V.M.; Altmann, D.R.; Miszkiel, K.A.; Thompson, A.J.; Miller, D.H. Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 64, 247–254. [Google Scholar] [CrossRef]
- Levin, M.C.; Jackson, W.C. Developing a therapeutic plan for treating MS: Evidence for new treatments. J. Clin. Psychiatry 2014, 75, e34. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C. Demyelinating Disorders. In The Merck Manual Professional Version; Porter, R., Ed.; Merck & Co., Inc.: Kenilworth, NJ, USA, 2018. [Google Scholar]
- Heidker, R.M.; Emerson, M.R.; LeVine, S.M. Metabolic pathways as possible therapeutic targets for progressive multiple sclerosis. Neural Regen. Res. 2017, 12, 1262. [Google Scholar] [PubMed]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Black, J.A.; Newcombe, J.; Trapp, B.D.; Waxman, S.G. Sodium channel expression within chronic multiple sclerosis plaques. J. Neuropathol. Exp. Neurol. 2007, 66, 828–837. [Google Scholar] [CrossRef]
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1. 2 and Nav1. 6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B. Regulated protein aggregation: Stress granules and neurodegeneration. Mol. Neurodegener. 2012, 7, 56. [Google Scholar] [CrossRef]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef]
- Aulas, A.; Lyons, S.M.; Fay, M.M.; Anderson, P.; Ivanov, P. Nitric oxide triggers the assembly of “type II” stress granules linked to decreased cell viability. Cell Death Dis. 2018, 9, 1129. [Google Scholar] [CrossRef]
- Reineke, L.C.; Neilson, J.R. Differences between acute and chronic stress granules, and how these differences may impact function in human disease. Biochem. Pharmacol. 2019, 162, 123–131. [Google Scholar] [CrossRef]
- Reineke, L.C.; Cheema, S.A.; Dubrulle, J.; Neilson, J.R. Chronic starvation induces noncanonical pro-death stress granules. J. Cell Sci. 2018, 131, jcs220244. [Google Scholar] [CrossRef]
- Bentmann, E.; Haass, C.; Dormann, D. Stress granules in neurodegeneration–lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBs J. 2013, 280, 4348–4370. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R. mRNP granules: Assembly, function, and connections with disease. RNA Biol. 2014, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Libner, C.D.; Salapa, H.E.; Hutchinson, C.; Lee, S.; Levin, M.C. Antibodies to the RNA Binding Protein Heterogeneous Nuclear Ribonucleoprotein A1 Contribute to Neuronal Cell Loss in an Animal Model of Multiple Sclerosis. J. Comp. Neurol. 2020, 528, 1704–1724. [Google Scholar] [CrossRef]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Salapa, H.E.; Libner, C.D.; Levin, M.C. Dysfunctional RNA-binding protein biology and neurodegeneration in experimental autoimmune encephalomyelitis in female mice. J. Neurosci. Res. 2020, 98, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Salapa, H.E.; Johnson, C.; Hutchinson, C.; Popescu, B.F.; Levin, M.C. Dysfunctional RNA binding proteins and stress granules in multiple sclerosis. J. Neuroimmunol. 2018, 324, 149–156. [Google Scholar] [CrossRef]
- Hock, E.-M.; Maniecka, Z.; Hruska-Plochan, M.; Reber, S.; Laferriere, F.; Mk, S.S.; Ederle, H.; Gittings, L.; Pelkmans, L.; Dupuis, L. Hypertonic stress causes cytoplasmic translocation of neuronal, but not astrocytic, FUS due to impaired transportin function. Cell Rep. 2018, 24, 987–1000. [Google Scholar] [CrossRef]
- Lee, S.; Levin, M. Novel Somatic Single Nucleotide Variants within the RNA Binding Protein hnRNP A1 in Multiple Sclerosis Patients. F1000Research 2014, 3, 132. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White Iii, C.L.; Bigio, E.H.; Caselli, R. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 63, 535–538. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Zhang, Y.-J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.-F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.P. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. Plos Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef] [PubMed]
- Salapa, H.E.; Hutchinson, C.; Popescu, B.F.; Levin, M.C. Neurons in multiple sclerosis cortex show features of RNA binding protein dysfunction. Ann. Clin. Transl. Neurol. 2020. [Google Scholar]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Masaki, K.; Sonobe, Y.; Ghadge, G.; Pytel, P.; Lépine, P.; Pernin, F.; Cui, Q.-L.; Antel, J.P.; Zandee, S.; Prat, A. RNA-binding protein altered expression and mislocalization in MS. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef]
- Lee, S.; Xu, L.; Shin, Y.; Gardner, L.; Hartzes, A.; Dohan, F.C.; Raine, C.; Homayouni, R.; Levin, M.C. A potential link between autoimmunity and neurodegeneration in immune-mediated neurological disease. J. Neuroimmunol. 2011, 235, 56–69. [Google Scholar] [CrossRef]
- Levin, M.C.; Lee, S.; Gardner, L.A.; Shin, Y.; Douglas, J.N.; Groover, C.J. Pathogenic mechanisms of neurodegeneration based on the phenotypic expression of progressive forms of immune-mediated neurologic disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 175. [Google Scholar] [CrossRef]
- Lee, S.; Salapa, H.E.; Levin, M.C. Localization of near-infrared labeled antibodies to the central nervous system in experimental autoimmune encephalomyelitis. PLoS ONE 2019, 14, e0212357. [Google Scholar] [CrossRef] [PubMed]
- Salapa, H.; Lee, S.; Shin, Y.; Levin, M. Contribution of the degeneration of the neuro-axonal unit to the pathogenesis of multiple sclerosis. Brain Sci. 2017, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C.; Lee, S.; Gardner, L.A.; Shin, Y.; Douglas, J.N.; Salapa, H. Autoantibodies to heterogeneous nuclear ribonuclear protein A1 (hnRNPA1) cause altered ‘ribostasis’ and neurodegeneration; the legacy of HAM/TSP as a model of progressive multiple sclerosis. J. Neuroimmunol. 2017, 304, 56–62. [Google Scholar] [CrossRef]
- Douglas, J.N.; Gardner, L.A.; Salapa, H.E.; Levin, M.C. Antibodies to the RNA binding protein heterogeneous nuclear ribonucleoprotein A1 colocalize to stress granules resulting in altered RNA and protein levels in a model of neurodegeneration in multiple sclerosis. J. Clin. Cell. Immunol. 2016, 7, 402. [Google Scholar]
- Douglas, J.N.; Gardner, L.A.; Salapa, H.E.; Lalor, S.J.; Lee, S.; Segal, B.M.; Sawchenko, P.E.; Levin, M.C. Antibodies to the RNA-binding protein hnRNP A1 contribute to neurodegeneration in a model of central nervous system autoimmune inflammatory disease. J. Neuroinflamm. 2016, 13, 178. [Google Scholar] [CrossRef]
- Rizzo, M.A.; Hadjimichael, O.C.; Preiningerova, J.; Vollmer, T.L. Prevalence and treatment of spasticity reported by multiple sclerosis patients. Mult. Scler. J. 2004, 10, 589–595. [Google Scholar] [CrossRef]
- Barnes, M.P.; Kent, R.M.; Semlyen, J.K.; McMullen, K.M. Spasticity in multiple sclerosis. Neurorehabilit. Neural Repair 2003, 17, 66–70. [Google Scholar] [CrossRef]