Lipopolysaccharides (LPSs) are bacterial surface glycolipids, produced by Gram-negative bacteria. LPS is known to determine acute inflammatory reactions, particularly in the context of sepsis. However, LPS can also trigger chronic inflammation. In this case, the source of LPS is not an external infection, but rather an increase in endogenous production, which is usually sustained by gut microbiota (GM), and LPS contained in food. The first site in which LPS can exert its inflammatory action is the gut: both GM and gut-associated lymphoid tissue (GALT) are influenced by LPS and shift towards an inflammatory pattern. The changes in GM and GALT induced by LPS are quite similar to the ones seen in IBD: GM loses diversity, while GALT T regulatory (Tregs) lymphocytes are reduced in number, with an increase in Th17 and Th1 lymphocytes. Additionally, the innate immune system is triggered, through the activation of toll-like receptor (TLR)-4, while the epithelium is directly damaged, further triggering inflammation.
- inflammation
- LPS
- microbiota
- Crohn’s disease
- ulcerative colitis
- IBD
- Th17
- Tregs
- metabolic endotoxemia
1. Introduction
2. LPS and IBD, a Complex Crosstalk
2.1. LPS and the Immune System
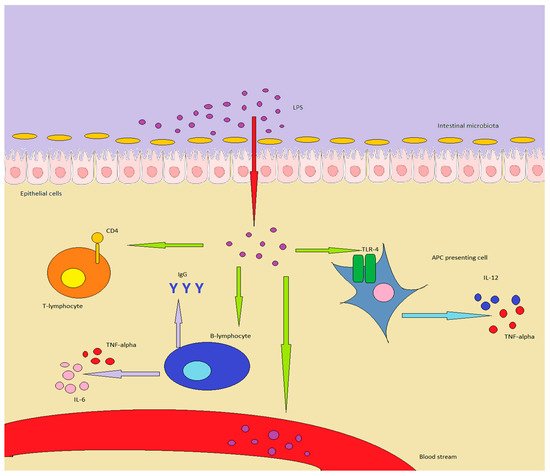
2.2. LPS and Microbiota
2.3. LPS and IBD, What Do We Know
This entry is adapted from the peer-reviewed paper 10.3390/ijms22126242
References
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8.
- Diks, S.H.; Richel, D.J.; Peppelenbosch, M.P. LPS signal transduction: The picture is becoming more complex. Curr. Top. Med. Chem. 2004, 4, 1115–1126.
- Yang, J.; Zhao, Y.; Shao, F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 2015, 32, 78–83.
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11, 594150.
- Ramendra, R.; Isnard, S.; Mehraj, V.; Chen, J.; Zhang, Y.; Finkelman, M.; Routy, J.P. Circulating LPS and (1→3)-β-D-Glucan: A Folie à Deux Contributing to HIV-Associated Immune Activation. Front. Immunol. 2019, 10, 465.
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193.
- Hurley, J.C.; Guidet, B.; Offenstadt, G.; Maury, E. Endotoxemia and mortality prediction in ICU and other settings: Underlying risk and co-detection of gram negative bacteremia are confounders. Crit. Care 2012, 16, R148.
- Napier, B.A.; Andres-Terre, M.; Massis, L.M.; Hryckowian, A.J.; Higginbottom, S.K.; Cumnock, K.; Casey, K.M.; Haileselassie, B.; Lugo, K.A.; Schneider, D.S.; et al. Western diet regulates immune status and the response to LPS-driven sepsis independent of diet-associated microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 3688–3694.
- Suzuki, K. Chronic Inflammation as an Immunological Abnormality and Effectiveness of Exercise. Biomolecules 2019, 9, 223.
- Zhang, Y.; Li, X.; Luo, Z.; Ma, L.; Zhu, S.; Wang, Z.; Wen, J.; Cheng, S.; Gu, W.; Lian, Q.; et al. ECM1 is an essential factor for the determination of M1 macrophage polarization in IBD in response to LPS stimulation. Proc. Natl. Acad. Sci. USA 2020, 117, 3083–3092.
- Bian, Y.; Dong, Y.; Sun, J.; Sun, M.; Hou, Q.; Lai, Y.; Zhang, B. Protective Effect of Kaempferol on LPS-Induced Inflammation and Barrier Dysfunction in a Coculture Model of Intestinal Epithelial Cells and Intestinal Microvascular Endothelial Cells. J. Agric. Food Chem. 2020, 68, 160–167.
- Heinbockel, L.; Weindl, G.; Martinez-de-Tejada, G.; Correa, W.; Sanchez-Gomez, S.; Bárcena-Varela, S.; Goldmann, T.; Garidel, P.; Gutsmann, T.; Brandenburg, K. Inhibition of Lipopolysaccharide- and Lipoprotein-Induced Inflammation by Antitoxin Peptide Pep19-2.5. Front. Immunol. 2018, 9, 1704.
- Jaffer, U.; Wade, R.G.; Gourlay, T. Cytokines in the systemic inflammatory response syndrome: A review. HSR Proc. Intensive Care Cardiovasc. Anesth. 2010, 2, 161–175.
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316.
- Rahhal, R.M.; Vanden Bush, T.J.; McLendon, M.K.; Apicella, M.A.; Bishop, G.A. Differential effects of Francisella tularensis lipopolysaccharide on B lymphocytes. J. Leukoc. Biol. 2007, 82, 813–820.
- Vazquez-Torres, A.; Vallance, B.A.; Bergman, M.A.; Finlay, B.B.; Cookson, B.T.; Jones-Carson, J.; Fang, F.C. Toll-Like Receptor 4 Dependence of Innate and Adaptive Immunity to Salmonella: Importance of the Kupffer Cell Network. J. Immunol. 2004, 172, 6202–6208.
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front. Pharmacol. 2017, 8, 278.
- Ding, F.; Fu, Z.; Liu, B. Lipopolysaccharide Exposure Alleviates Asthma in Mice by Regulating Th1/Th2 and Treg/Th17 Balance. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 3220–3229.
- Straub, T.; Freudenberg, M.A.; Schleicher, U.; Bogdan, C.; Gasteiger, G.; Pircher, H. Bacterial coinfection restrains antiviral CD8 T-cell response via LPS-induced inhibitory NK cells. Nat. Commun. 2018, 9, 4117.
- Bannerman, D.D.; Goldblum, S.E. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L899–L914.
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.-M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853.
- Orivuori, L.; Mustonen, K.; de Goffau, M.C.; Hakala, S.; Paasela, M.; Roduit, C.; Dalphin, J.C.; Genuneit, J.; Lauener, R.; Riedler, J.; et al. High level of fecal calprotectin at age 2 months as a marker of intestinal inflammation predicts atopic dermatitis and asthma by age 6. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2015, 45, 928–939.
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277.
- Wassenaar, T.M.; Zimmermann, K. Lipopolysaccharides in Food, Food Supplements, and Probiotics: Should We be Worried? Eur. J. Microbiol. Immunol. 2018, 8, 63–69.
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511.
- Lindenberg, F.C.B.; Ellekilde, M.; Thörn, A.C.; Kihl, P.; Larsen, C.S.; Hansen, C.H.F.; Metzdorff, S.B.; Aalbæk, B.; Hansen, A.K. Dietary LPS traces influences disease expression of the diet-induced obese mouse. Res. Vet. Sci. 2019, 123, 195–203.
- Netto Candido, T.L.; Bressan, J.; Alfenas, R.C.G. Dysbiosis and metabolic endotoxemia induced by high-fat diet. Nutr. Hosp. 2018, 35, 1432–1440.
- Matsuoka, K.; Kanai, T. The gut microbiota and inflammatory bowel disease. Semin. Immunopathol. 2015, 37, 47–55.
- Wang, J.; Chen, W.-D.; Wang, Y.-D. The Relationship Between Gut Microbiota and Inflammatory Diseases: The Role of Macrophages. Front. Microbiol. 2020, 11, 1065.
- Franza, L.; Carusi, V.; Altamura, S.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Conti, P.; Pandolfi, F. Interrelationship between inflammatory cytokines (IL-1, IL-6, IL-33, IL-37) and acquired immunity. J. Biol. Regul. Homeost. Agents 2019, 33, 1321–1326.
- Pandolfi, F.; Franza, L.; Carusi, V.; Altamura, S.; Andriollo, G.; Nucera, E. Interleukin-6 in Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 5238.
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792.
- Wang, J.W.; Pan, Y.B.; Cao, Y.Q.; Wang, C.; Jiang, W.D.; Zhai, W.F.; Lu, J.G. Loganin alleviates LPS-activated intestinal epithelial inflammation by regulating TLR4/NF-κB and JAK/STAT3 signaling pathways. Kaohsiung J. Med. Sci. 2020, 36, 257–264.
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51.
- Førland, D.T.; Johnson, E.; Saetre, L.; Lyberg, T.; Lygren, I.; Hetland, G. Effect of an extract based on the medicinal mushroom Agaricus blazei Murill on expression of cytokines and calprotectin in patients with ulcerative colitis and Crohn’s disease. Scand. J. Immunol. 2011, 73, 66–75.
- D’Amico, F.; Rubin, D.T.; Kotze, P.G.; Magro, F. International consensus on methodological issues in standardization of fecal calprotectin measurement in inflammatory bowel diseases. UEG J. 2021, 9, 451–460.
- Jaworska, K.; Konop, M.; Bielinska, K.; Hutsch, T.; Dziekiewicz, M.; Banaszkiewicz, A.; Ufnal, M. Inflammatory bowel disease is associated with increased gut-to-blood penetration of short-chain fatty acids: A new, non-invasive marker of a functional intestinal lesion. Exp. Physiol. 2019, 104, 1226–1236.
- Kiecolt-Glaser, J.K.; Wilson, S.J.; Bailey, M.L.; Andridge, R.; Peng, J.; Jaremka, L.M.; Fagundes, C.P.; Malarkey, W.B.; Laskowski, B.; Belury, M.A. Marital distress, depression, and a leaky gut: Translocation of bacterial endotoxin as a pathway to inflammation. Psychoneuroendocrinology 2018, 98, 52–60.