Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Alcoholic liver disease (ALD) is a globally prevalent chronic liver disease caused by chronic or binge consumption of alcohol. Metabolites and byproducts generated during alcohol metabolism cause liver damage, leading to ALD via several mechanisms, such as impairing lipid metabolism, intensifying inflammatory reactions, and inducing fibrosis.
- alcoholic liver disease
- alcohol metabolism
1. Introduction
Alcohol has long been closely related to human culture, used as a drug, and considered an indulgence [1][2]. As the consumption of alcoholic beverages increases, it contributes to the significant elevation of morbidity and mortality worldwide [3][4]. In 2018, the World Health Organization (WHO) estimated that alcohol consumption is responsible for three million deaths worldwide annually, accounting for nearly 14% of the total mortality in people aged 20 to 40 years old [5]. In addition to social and psychiatric problems, more than 200 diseases that affect several organs, such as the brain, heart, gastrointestinal tract, and liver, are related to habitual alcohol consumption [6][7][8]. Among these alcohol-damaged organs, the liver is specifically susceptible to damage because the liver is the primary site of alcohol metabolism in the body [9][10]. Alcohol metabolism generates products that damage the liver, resulting in alcoholic liver disease (ALD), a main cause of chronic liver disease [4][9][10][11]. ALD encompasses a broad spectrum of conditions, including alcoholic fatty liver (simple steatosis), alcoholic hepatitis, alcoholic cirrhosis, and liver cancer [4][11][12][13]. Alcoholic fatty liver is defined by fat accumulation in hepatocytes without substantial inflammation, or hepatic fibrosis, and is observed in up to 90% of heavy drinkers [9][11][12][13][14]. Heavy long-term alcohol consumption accelerates the transition of alcoholic fatty liver into alcoholic hepatitis, which is characterized by steatosis, ballooning hepatocytes, and infiltration of neutrophils with or without fibrosis [11][12][13][14][15][16]. Alcoholic cirrhosis is a more severe form of ALD and is defined by disorganized liver architecture with fibrosis [11][12][13][14][15][16].
2. Alcohol Metabolism
Ingested alcohol is absorbed through the stomach and intestines. Less than 10% of absorbed alcohol is excreted in breath, sweat, and urine. This means that more than 90% of the absorbed alcohol circulates through the body and is eventually transported to the liver via the portal vein [10][17]. Due to the high levels of alcohol metabolizing enzymes in the liver, the liver plays a major role in alcohol metabolism [10]. In the liver, alcohol is metabolized by oxidative and non-oxidative pathways (Figure 1) [18][19]. The oxidative pathway is the major pathway for alcohol metabolism and is composed of two steps. First, alcohol is oxidized to acetaldehyde by alcohol dehydrogenase (ADH), a primary enzyme that converts alcohol to acetaldehyde [20]. Excess alcohol consumption increases the expression and activity of cytochrome P450 2E1 (CYP2E1), not ADH. Activated CYP2E1 promotes the production of acetaldehyde through the formation of reactive oxygen species (ROS) [21][22]. In addition, peroxisomal catalase breaks down alcohol to acetaldehyde, but its action is considered a minor pathway because of its small contribution to alcohol digestion [10][19]. The second step in the oxidative pathway involves the rapid conversion of acetaldehyde to acetate by aldehyde dehydrogenase (ALDH). Acetate is metabolized into carbon dioxide (CO2), fatty acids (FAs), and water (H2O) in peripheral tissues, not the liver [10][19]. The nonoxidative pathway accounts for a minor portion of alcohol metabolism in quantitative terms [23][24][25]. A small amount of alcohol is nonoxidatively conjugated to various endogenous metabolites by different enzymes. For example, enzymatic esterification of alcohol with FAs forms fatty acid ethyl ester (FAEE) and phospholipase D (PLD) catalyzes transphosphatidylation of phosphatidylcholine with ethanol to form phosphatidylethanol (PEth). In addition, alcohol conjugated to glucuronic acid and sulfate generates ethyl glucuronide (EtG) and ethyl sulfate (EtS), respectively [23][24][25].
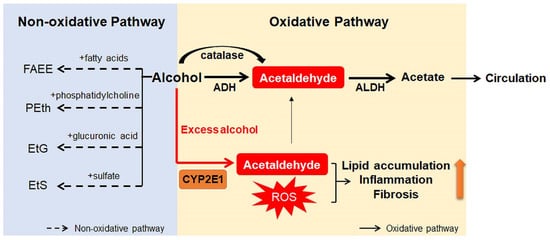
Figure 1. Scheme for alcohol metabolism in the liver. In the liver, alcohol is metabolized by the oxidative and non-oxidative pathway. In the oxidative pathway, the major pathway of alcohol digestion, alcohol is oxidized to acetaldehyde by various enzymes including alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1), and catalase. Then, acetaldehyde is broken down into acetate, which is excreted out of the liver. Especially when excessive alcohol is consumed, CYP2E1 is activated and promotes formation of reactive oxygen species (ROS). The non-oxidative pathway accounts for a small amount of alcohol metabolism. Various enzymes nonoxidatively conjugates alcohol with different endogenous metabolites, producing fatty acid ethyl ester (FAEE), phosphatidylethanol (PEth), ethyl glucuronide (EtG), and ethyl sulfate (EtS). The byproducts generated during alcohol metabolism injure the liver by increasing lipid accumulation, inflammation, and fibrosis. Especially, acetaldehyde, the first metabolite of alcohol metabolism, is well known for toxic compounds. ROS, which are generated by activation of CYP2E1, are also considered as one of the major contributors of liver damage. In addition, acetate and non-oxidative metabolites are known to damage the liver.
Products generated during alcohol metabolism damage the liver and act as a driving force of ALD progression from alcoholic steatosis to alcoholic cirrhosis [26][27]. The most well-known toxic compound produced by alcohol metabolism is acetaldehyde [28]. Acetaldehyde interacts directly with DNA and causes point mutations and chromosomal damage. It also binds to a variety of proteins to form acetaldehyde adducts, which distorts liver function and structure [29][30]. These protein adducts upregulate CYP2E1 expression and enhance oxidative stress [31][32]. In addition, Holstege et al. [33] demonstrated that protein adducts contribute to lipid accumulation, inflammation, and fibrosis, playing a key role in the pathogenesis of various stages of ALD. Acetate has been reported to increase portal blood flow by circulating in the bloodstream, although it is less toxic than acetaldehyde [34]. Nonoxidative pathway-derived metabolites, including PEth and FAEE, are also known to cause alcohol toxicity, but the mechanisms have not yet been determined [23][35].
3. Oxidative Ethanol Metabolites Are Involved in ALD Pathogenesis
Oxidative ethanol-derived metabolites exert a broad spectrum of damage in the liver, ranging from lipid accumulation in hepatocytes to inflammation, fibrosis, and carcinogenesis (Figure 2). Excessive lipid accumulation in hepatocytes results in massive hepatocyte death, which triggers pro-inflammatory and pro-fibrogenic responses, increasing the risk of liver cancer [36]. Because there is a dearth of information about ALD pathogenesis exerted by non-oxidative ethanol metabolism, this section reviews the mechanisms underlying oxidative ethanol metabolite-mediated hepatotoxicity caused by excess alcohol exposure [37].
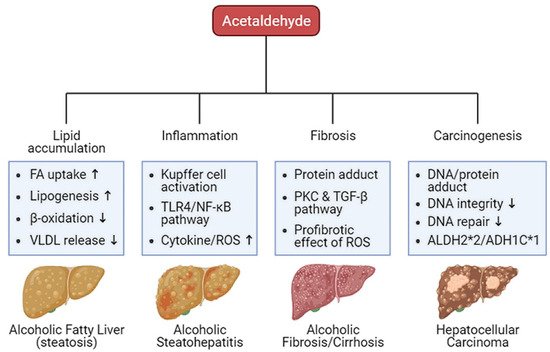
Figure 2. Harmful effects of acetaldehyde on ALD. Acetaldehyde, one of the oxidative ethanol-derived metabolites, exerts a broad spectrum of damage to the liver, ranging from lipid accumulation in hepatocytes to inflammation, fibrosis, and carcinogenesis. The hepatocytes accumulate lipids by the lipid oversupply (increase of fatty acid (FA) uptake to the liver and de novo lipogenesis in the liver) and/or the impaired pathway of lipid clearance (decrease of mitochondrial β-oxidation and secretion of excessive lipids in very-low density lipoprotein (VLDL)). In addition, acetaldehyde activates Kupffer cells, the liver-resident macrophage, to release reactive oxygen species (ROS) and cytokines that recruit other immune cells. When exposed to acetaldehyde, Kupffer cells activate the toll-like receptor 4 (TLR4)-mediated nuclear factor (NF)-κB signaling pathway, triggering inflammatory responses. The acetaldehyde-protein adducts promote the collagen production by activated hepatic stellate cells (HSCs) via the protein kinase C (PKC) and the TGF-β signaling pathway. ROS-mediated oxidative stress also accelerates liver fibrosis. Moreover, protein and DNA adducts with acetaldehyde cause hepatic carcinogenesis. They weaken DNA integrity and interfere with DNA repair system, increasing carcinogenic DNA mutation. Genetic variations in the alcohol-metabolizing enzymes, such as acetaldehyde dehydrogenase (ALDH)2*2 and alcohol dehydrogenase (ADH)1C*1 alleles, suppress the activity of the enzymes, enhancing the amount of acetaldehyde.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22115717
References
- Sudhinaraset, M.; Wigglesworth, C.; Takeuchi, D.T. Social and Cultural Contexts of Alcohol Use: Influences in a Social-Ecological Framework. Alcohol Res. 2016, 38, 35–45.
- Khaderi, S.A. Introduction: Alcohol and Alcoholism. Clin. Liver Dis. 2019, 23, 1–10.
- Stockwell, T.; Zhao, J.; Panwar, S.; Roemer, A.; Naimi, T.; Chikritzhs, T. Do “Moderate” Drinkers Have Reduced Mortality Risk? A Systematic Review and Meta-Analysis of Alcohol Consumption and All-Cause Mortality. J. Stud. Alcohol Drugs 2016, 77, 185–198.
- Zakhari, S.; Li, T.K. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology 2007, 46, 2032–2039.
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9.
- Shield, K.D.; Parry, C.; Rehm, J. Chronic diseases and conditions related to alcohol use. Alcohol Res. 2013, 35, 155–173.
- Rehm, J.; Baliunas, D.; Borges, G.L.; Graham, K.; Irving, H.; Kehoe, T.; Parry, C.D.; Patra, J.; Popova, S.; Poznyak, V.; et al. The relation between different dimensions of alcohol consumption and burden of disease: An overview. Addiction 2010, 105, 817–843.
- Corrao, G.; Bagnardi, V.; Zambon, A.; La Vecchia, C. A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev. Med. 2004, 38, 613–619.
- Lieber, C.S. Alcoholic liver disease: New insights in pathogenesis lead to new treatments. J. Hepatol. 2000, 32, 113–128.
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685.
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585.
- Altamirano, J.; Bataller, R. Alcoholic liver disease: Pathogenesis and new targets for therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 491–501.
- Schwartz, J.M.; Reinus, J.F. Prevalence and natural history of alcoholic liver disease. Clin. Liver Dis. 2012, 16, 659–666.
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19.
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017, 38, 147–161.
- Askgaard, G.; Grønbæk, M.; Kjær, M.S.; Tjønneland, A.; Tolstrup, J.S. Alcohol drinking pattern and risk of alcoholic liver cirrhosis: A prospective cohort study. J. Hepatol. 2015, 62, 1061–1067.
- Holford, N.H. Clinical pharmacokinetics of ethanol. Clin. Pharm. 1987, 13, 273–292.
- Lieber, C.S. Metabolism of alcohol. Clin. Liver Dis. 2005, 9, 1–35.
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254.
- Lieber, C.S. Ethanol metabolism, cirrhosis and alcoholism. Clin. Chim. Acta. 1997, 257, 59–84.
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738.
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398.
- Heier, C.; Xie, H.; Zimmermann, R. Nonoxidative ethanol metabolism in humans-from biomarkers to bioactive lipids. IUBMB Life 2016, 68, 916–923.
- Maenhout, T.M.; De Buyzere, M.L.; Delanghe, J.R. Non-oxidative ethanol metabolites as a measure of alcohol intake. Clin. Chim. Acta. 2013, 415, 322–329.
- Laposata, E.A.; Lange, L.G. Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 1986, 231, 497–499.
- Lieber, C.S. Hepatic and metabolic effects of ethanol: Pathogenesis and prevention. Ann. Med. 1994, 26, 325–330.
- Lieber, C.S. Mechanism of ethanol induced hepatic injury. Pharmacol. Ther. 1990, 46, 1–41.
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185.
- Tuma, D.J.; Casey, C.A. Dangerous byproducts of alcohol breakdown--focus on adducts. Alcohol Res. Health 2003, 27, 285–290.
- Israel, Y.; Hurwitz, E.; Niemelä, O.; Arnon, R. Monoclonal and polyclonal antibodies against acetaldehyde-containing epitopes in acetaldehyde-protein adducts. Proc. Natl. Acad. Sci. USA 1986, 83, 7923–7927.
- Niemelä, O.; Parkkila, S.; Pasanen, M.; Iimuro, Y.; Bradford, B.; Thurman, R.G. Early alcoholic liver injury: Formation of protein adducts with acetaldehyde and lipid peroxidation products, and expression of CYP2E1 and CYP3A. Alcohol Clin. Exp. Res. 1998, 22, 2118–2124.
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 2003, 27, 277–284.
- Holstege, A.; Bedossa, P.; Poynard, T.; Kollinger, M.; Chaput, J.C.; Houglum, K.; Chojkier, M. Acetaldehyde-modified epitopes in liver biopsy specimens of alcoholic and nonalcoholic patients: Localization and association with progression of liver fibrosis. Hepatology 1994, 19, 367–374.
- Israel, Y.; Orrego, H.; Carmichael, F.J. Acetate-mediated effects of ethanol. Alcohol Clin. Exp. Res. 1994, 18, 144–148.
- Seth, D.; Haber, P.S.; Syn, W.K.; Diehl, A.M.; Day, C.P. Pathogenesis of alcohol-induced liver disease: Classical concepts and recent advances. J. Gastroenterol. Hepatol. 2011, 26, 1089–1105.
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194.
- Dinis-Oliveira, R.J. Oxidative and Non-Oxidative Metabolomics of Ethanol. Curr. Drug Metab. 2016, 17, 327–335.
This entry is offline, you can click here to edit this entry!