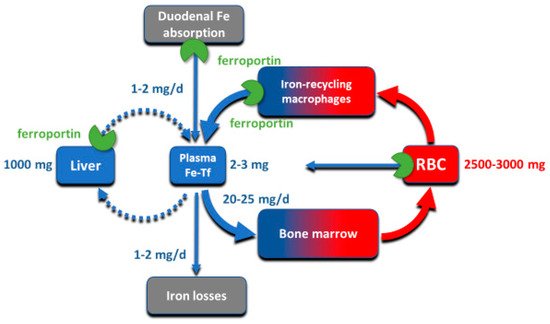
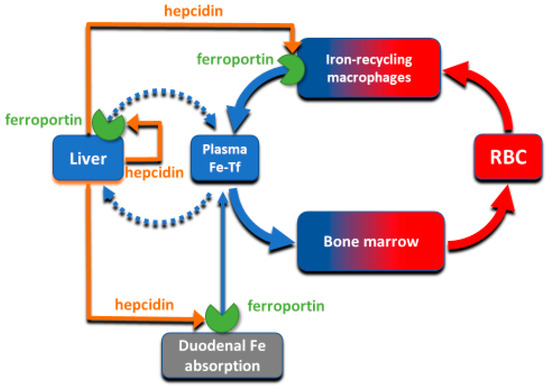
Despite its abundance in the environment, iron is poorly bioavailable and subject to strict conservation and internal recycling by most organisms. In vertebrates, the stability of iron concentration in plasma and extracellular fluid, and the total body iron content are maintained by the interaction of the iron-regulatory peptide hormone hepcidin with its receptor and cellular iron exporter ferroportin (SLC40a1). Ferroportin exports iron from duodenal enterocytes that absorb dietary iron, from iron-recycling macrophages in the spleen and the liver, and from iron-storing hepatocytes. Hepcidin blocks iron export through ferroportin, causing hypoferremia. During iron deficiency or after hemorrhage, hepcidin decreases to allow iron delivery to plasma through ferroportin, thus promoting compensatory erythropoiesis. As a host defense mediator, hepcidin increases in response to infection and inflammation, blocking iron delivery through ferroportin to blood plasma, thus limiting iron availability to invading microbes. Genetic diseases that decrease hepcidin synthesis or disrupt hepcidin binding to ferroportin cause the iron overload disorder hereditary hemochromatosis.
- iron deficiency
- iron overload
- hemochromatosis
- anemia
- metal transport
1. The Roles of Iron
2. Iron Compartments and Flows
3. Molecular Basis of Systemic Iron Homeostasis
4. Ferroportin Structure
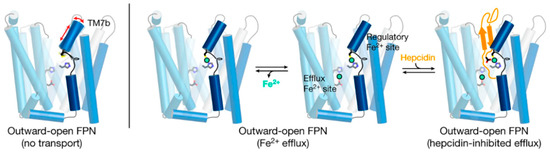
5. Ferroportin Interactions with Hepcidin
This entry is adapted from the peer-reviewed paper 10.3390/ijms22126493
References
- Fillet, G.; Beguin, Y.; Baldelli, L. Model of reticuloendothelial iron metabolism in humans: Abnormal behavior in idiopathic hemochromatosis and in inflammation. Blood 1989, 74, 844–851.
- Morgan, E.H.; Walters, M.N. Iron storage in human disease: Fractionation of hepatic and splenic iron into ferritin and haemosiderin with histochemical correlations. J. Clin. Pathol. 1963, 16, 101–107.
- Finch, C. Regulators of iron balance in humans. Blood 1994, 84, 1697–1702.
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093.
- Ganz, T.; Olbina, G.; Girelli, D.; Nemeth, E.; Westerman, M. Immunoassay for human serum hepcidin. Blood 2008, 112, 4292–4297.
- Troutt, J.S.; Rudling, M.; Persson, L.; Stahle, L.; Angelin, B.; Butterfield, A.M.; Schade, A.E.; Cao, G.; Konrad, R.J. Circulating human hepcidin-25 concentrations display a diurnal rhythm, increase with prolonged fasting, and are reduced by growth hormone administration. Clin. Chem. 2012, 58, 1225–1232.
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781.
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309.
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912.
- Canonne-Hergaux, F.; Donovan, A.; Delaby, C.; Wang, H.J.; Gros, P. Comparative studies of duodenal and macrophage ferroportin proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G156–G163.
- Taniguchi, R.; Kato, H.E.; Font, J.; Deshpande, C.N.; Wada, M.; Ito, K.; Ishitani, R.; Jormakka, M.; Nureki, O. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat. Commun. 2015, 6, 8545.
- Mitchell, C.J.; Shawki, A.; Ganz, T.; Nemeth, E.; Mackenzie, B. Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc. Am. J. Physiol. Cell Physiol. 2013, 306, C450–C459.
- Deshpande, C.N.; Ruwe, T.A.; Shawki, A.; Xin, V.; Vieth, K.R.; Valore, E.V.; Qiao, B.; Ganz, T.; Nemeth, E.; Mackenzie, B.; et al. Calcium is an essential cofactor for metal efflux by the ferroportin transporter family. Nat. Commun. 2018, 9, 3075.
- Billesbolle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 2020, 586, 807–811.
- Philpott, C.C.; Patel, S.J.; Protchenko, O. Management versus miscues in the cytosolic labile iron pool: The varied functions of iron chaperones. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118830.
- Yanatori, I.; Richardson, D.R.; Imada, K.; Kishi, F. Iron export through the transporter ferroportin 1 is modulated by the iron chaperone PCBP2. J. Biol. Chem. 2016, 291, 17303–17318.
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910.
- Rivera, S.; Liu, L.; Nemeth, E.; Gabayan, V.; Sorensen, O.E.; Ganz, T. Hepcidin excess induces the sequestration of iron and exacerbates tumor-associated anemia. Blood 2005, 105, 1797–1802.
- Ross, S.L.; Tran, L.; Winters, A.; Lee, K.J.; Plewa, C.; Foltz, I.; King, C.; Miranda, L.P.; Allen, J.; Beckman, H.; et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metab. 2012, 15, 905–917.
- Qiao, B.; Sugianto, P.; Fung, E.; del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924.
- Jiang, L.; Wang, J.; Wang, K.; Wang, H.; Wu, Q.; Yang, C.; Yu, Y.; Ni, P.; Zhong, Y.; Song, Z.; et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood 2021.
- Link, C.; Knopf, J.D.; Marques, O.; Lemberg, M.K.; Muckenthaler, M.U. The role of cellular iron deficiency in controlling iron export. Biochim. Biophys. Acta Gen. Subj. 2020, 1865, 129829.
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Investig. 2011, 121, 4880–4888.
- Manolova, V.; Nyffenegger, N.; Flace, A.; Altermatt, P.; Varol, A.; Doucerain, C.; Sundstrom, H.; Durrenberger, F. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of beta-thalassemia. J. Clin. Investig. 2019, 130, 491–506.