Idiosyncratic drug-induced liver injury (DILI) is a type of hepatic injury caused by an uncommon drug adverse reaction that can develop to conditions spanning from asymptomatic liver laboratory abnormalities to acute liver failure (ALF) and death.
- DILI
- oxidative stress
- pharmacology
- mitochondrial damage
1. Introduction
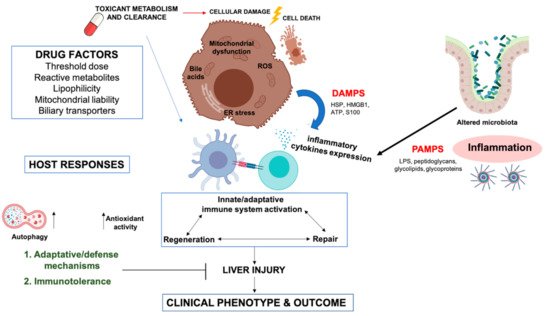
Drug-induced liver injury (DILI) is an adverse reaction caused by exposure to drugs and herbal medicines or other xenobiotics. Depending on the presumed mechanism of action of the causative drug, DILI is typically classified as intrinsic (direct) or idiosyncratic [1], although indirect injury is emerging as a third type [2]. Intrinsic DILI is related to the cytotoxic properties of the causative drug or its metabolite(s). In this case, liver injury is dose-dependent and predictable, and damage can be reproduced in animal models [3]. Acetaminophen (APAP) toxicity is the most common cause for this type of DILI [4][5][6][7]. In contrast, idiosyncratic DILI is mostly host-dependent, multifactorial and unpredictable, since it is determined by both the properties of the drug and its interaction with environmental and host factors [8]. Idiosyncratic DILI is usually not dose-dependent, although the exposure to a threshold dose in each susceptible individual is necessary [9][10]. Moreover, the delay between starting the drug and the onset of clinical signs of liver injury is another characteristic of idiosyncratic DILI. Indirect liver injury is caused by an indirect action of the drug on liver or immune system, and can induce a new liver condition or exacerbate a preexisting one, such as worsening of hepatitis B or C.
The lines that distinguish types of hepatotoxicity are blurred, and majority of drug-induced liver reactions are considered idiosyncratic. Indeed, this is an unresolved issue and rather an academic classification as research over the last years has demonstrated that there are host susceptibility factors that influence the risk of intrinsic damage and, on the contrary, for drugs that are believed to cause idiosyncratic liver damage, there might be a dose threshold. Therefore, unless stated otherwise, the term DILI is used for idiosyncratic drug-induced hepatotoxicity in this review.
2. Potential Mechanisms Involved in DILI Pathogenesis
The liver plays an important role in metabolizing drugs or exogenous toxicants, protecting the organism from potential toxic chemicals [11]. Bioactivation processes of parent drugs rendering reactive metabolites and the mechanisms involved in detoxification and excretion of xenobiotics (most of them under genetic control) are critical for understanding the mechanisms of DILI [12]. However, many different hypotheses have been proposed due to the multivariant nature of the disease [13].
2.1. Drug Factors
2.2. Metabolic Mechanisms
2.3. BSEP Inhibition
2.4. Activation of the Immune Response
It has been increasingly clear that the immune response during DILI is determinant, since the presence of T cells and immune system activation in patients with DILI have been observed [14][34][35]. The immune response consists of a hypersensitivity reaction which provokes an inflammatory response that involves the innate and the adaptive immune system. Different hypotheses have been suggested to explain drug-induced immune system activation [23].
2.4.1. The Hapten Hypothesis
2.4.2. The Danger Hypothesis
2.4.3. The Pharmacological Interaction (p-i) Hypothesis
2.4.4. The Altered Peptide Repertoire Hypothesis
2.4.5. The Multiple Determinant Hypothesis
2.4.6. The Inflammatory Stress Hypothesis
This hypothesis suggests that a potential inflammation occurring during drug treatment could interact with the action of the compound and produce liver injury [41]. Hepatic inflammation is often observed in DILI; therefore, it is suggested that DILI reactions could be unmasked by inflammation occurring during drug therapy. Inflammagens could bind to TLR or T-cell receptors (TCR), initiating the expression of inflammatory mediators. The inflammatory stress hypothesis has provided the first animal models in which liver injury is induced from different drugs associated with human DILI [55][56].
Moreover, a common mechanism of immune response involves activation of the inflammasome [57]. A recent study showed that the supernatant (presumably containing DAMPs) from the incubation of human hepatocytes with drugs that induce DILI activates the inflammasome in THP-1 cells, a macrophage cell line [58][59].
This entry is adapted from the peer-reviewed paper 10.3390/antiox10030390
References
- Roth, R.A.; Ganey, P.E. Intrinsic versus idiosyncratic drug-induced hepatotoxicity—Two villains or one? J. Pharmacol. Exp. Ther. 2010, 332, 692–697.
- Hoofnagle, J.H.; Bjornsson, E.S. Drug-Induced Liver Injury—Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273.
- Andrade, R.J.; Chalasani, N.; Bjornsson, E.S.; Suzuki, A.; Kullak-Ublick, G.A.; Watkins, P.B.; Devarbhavi, H.; Merz, M.; Lucena, M.I.; Kaplowitz, N.; et al. Drug-induced liver injury. Nat. Rev. Dis. Prim. 2019, 5, 58.
- Bernal, W.; Hyyrylainen, A.; Gera, A.; Audimoolam, V.K.; McPhail, M.J.; Auzinger, G.; Rela, M.; Heaton, N.; O’Grady, J.G.; Wendon, J.; et al. Lessons from look-back in acute liver failure? A single centre experience of 3300 patients. J. Hepatol. 2013, 59, 74–80.
- Gyamlani, G.G.; Parikh, C.R. Acetaminophen toxicity: Suicidal vs. accidental. Crit. Care 2002, 6, 155–159.
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiodt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372.
- Reuben, A.; Tillman, H.; Fontana, R.J.; Davern, T.; McGuire, B.; Stravitz, R.T.; Durkalski, V.; Larson, A.M.; Liou, I.; Fix, O.; et al. Outcomes in Adults With Acute Liver Failure Between 1998 and 2013: An Observational Cohort Study. Ann. Intern. Med. 2016, 164, 724–732.
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 2015, 63, 503–514.
- Carrascosa, M.F.; Salcines-Caviedes, J.R.; Lucena, M.I.; Andrade, R.J. Acute liver failure following atorvastatin dose escalation: Is there a threshold dose for idiosyncratic hepatotoxicity? J. Hepatol. 2015, 62, 751–752.
- Lammert, C.; Einarsson, S.; Saha, C.; Niklasson, A.; Bjornsson, E.; Chalasani, N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: Search for signals. Hepatology 2008, 47, 2003–2009.
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20.
- Andrade, R.J.; Robles, M.; Ulzurrun, E.; Lucena, M.I. Drug-induced liver injury: Insights from genetic studies. Pharmacogenomics 2009, 10, 1467–1487.
- Uetrecht, J. Mechanistic Studies of Idiosyncratic DILI: Clinical Implications. Front. Pharmacol. 2019, 10, 837.
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875.
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396.
- Knowles, S.R.; Uetrecht, J.; Shear, N.H. Idiosyncratic drug reactions: The reactive metabolite syndromes. Lancet 2000, 356, 1587–1591.
- Uetrecht, J. Idiosyncratic drug reactions: Current understanding. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 513–539.
- Stepan, A.F.; Walker, D.P.; Bauman, J.; Price, D.A.; Baillie, T.A.; Kalgutkar, A.S.; Aleo, M.D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410.
- Garcia-Cortes, M.; Robles-Diaz, M.; Stephens, C.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Drug induced liver injury: An update. Arch. Toxicol. 2020.
- Drug-Induced Liver Injury (DILI): Current Status and Future Directions for Drug Development and the Post-Market Setting; A Consensus by a CIOMS Working Group; Council for International Organizations of Medical Sciences (CIOMS): Geneva, Switzerland, 2020.
- Cubero, F.J.; Zoubek, M.E.; Hu, W.; Peng, J.; Zhao, G.; Nevzorova, Y.A.; Al Masaoudi, M.; Bechmann, L.P.; Boekschoten, M.V.; Muller, M.; et al. Combined Activities of JNK1 and JNK2 in Hepatocytes Protect Against Toxic Liver Injury. Gastroenterology 2016, 150, 968–981.
- Yuan, L.; Kaplowitz, N. Mechanisms of drug-induced liver injury. Clin. Liver Dis. 2013, 17, 507–518.
- Iorga, A.; Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int. J. Mol. Sci. 2017, 18, 1018.
- Walgren, J.L.; Mitchell, M.D.; Thompson, D.C. Role of metabolism in drug-induced idiosyncratic hepatotoxicity. Crit. Rev. Toxicol. 2005, 35, 325–361.
- Torres, S.; Baulies, A.; Insausti-Urkia, N.; Alarcon-Vila, C.; Fucho, R.; Solsona-Vilarrasa, E.; Nunez, S.; Robles, D.; Ribas, V.; Wakefield, L.; et al. Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure. Gastroenterology 2019, 157, 552–568.
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151.
- Morgan, R.E.; Trauner, M.; van Staden, C.J.; Lee, P.H.; Ramachandran, B.; Eschenberg, M.; Afshari, C.A.; Qualls, C.W., Jr.; Lightfoot-Dunn, R.; Hamadeh, H.K. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol. Sci. 2010, 118, 485–500.
- Stieger, B. Role of the bile salt export pump, BSEP, in acquired forms of cholestasis. Drug Metab. Rev. 2010, 42, 437–445.
- Whitington, P.F.; Freese, D.K.; Alonso, E.M.; Schwarzenberg, S.J.; Sharp, H.L. Clinical and biochemical findings in progressive familial intrahepatic cholestasis. J. Pediatr. Gastroenterol. Nutr. 1994, 18, 134–141.
- Fattinger, K.; Funk, C.; Pantze, M.; Weber, C.; Reichen, J.; Stieger, B.; Meier, P.J. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: A potential mechanism for hepatic adverse reactions. Clin. Pharmacol. Ther. 2001, 69, 223–231.
- Funk, C.; Ponelle, C.; Scheuermann, G.; Pantze, M. Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: In vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Mol. Pharmacol. 2001, 59, 627–635.
- Aleo, M.D.; Luo, Y.; Swiss, R.; Bonin, P.D.; Potter, D.M.; Will, Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014, 60, 1015–1022.
- Köck, K.; Ferslew, B.C.; Netterberg, I.; Yang, K.; Urban, T.J.; Swaan, P.W.; Stewart, P.W.; Brouwer, K.L. Risk factors for development of cholestatic drug-induced liver injury: Inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab. Dispos. 2014, 42, 665–674.
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63.
- Nagy, G.; Kardon, T.; Wunderlich, L.; Szarka, A.; Kiss, A.; Schaff, Z.; Bánhegyi, G.; Mandl, J. Acetaminophen induces ER dependent signaling in mouse liver. Arch. BioChem Biophys. 2007, 459, 273–279.
- Wuillemin, N.; Adam, J.; Fontana, S.; Krahenbuhl, S.; Pichler, W.J.; Yerly, D. HLA haplotype determines hapten or p-i T cell reactivity to flucloxacillin. J. Immunol. 2013, 190, 4956–4964.
- Pradeu, T.; Cooper, E.L. The danger theory: 20 years later. Front. Immunol. 2012, 3, 287.
- Pirmohamed, M.; Naisbitt, D.J.; Gordon, F.; Park, B.K. The danger hypothesis—Potential role in idiosyncratic drug reactions. Toxicology 2002, 181–182, 55–63.
- Burgdorf, S.; Kurts, C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr. Opin. Immunol. 2008, 20, 89–95.
- Mak, A.; Uetrecht, J. Immune mechanisms of idiosyncratic drug-induced liver injury. J. Clin. Transl. Res. 2017, 3, 145–156.
- Deng, X.; Luyendyk, J.P.; Ganey, P.E.; Roth, R.A. Inflammatory stress and idiosyncratic hepatotoxicity: Hints from animal models. Pharmacol. Rev. 2009, 61, 262–282.
- Li, J.; Uetrecht, J.P. The danger hypothesis applied to idiosyncratic drug reactions. Handb. Exp. Pharmacol. 2010, 493–509.
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218.
- Pichler, W.J. Pharmacological interaction of drugs with antigen-specific immune receptors: The p-i concept. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 301–305.
- Pichler, W.J.; Beeler, A.; Keller, M.; Lerch, M.; Posadas, S.; Schmid, D.; Spanou, Z.; Zawodniak, A.; Gerber, B. Pharmacological interaction of drugs with immune receptors: The p-i concept. Allergol. Int. 2006, 55, 17–25.
- Pichler, W.J. The p-i Concept: Pharmacological Interaction of Drugs With Immune Receptors. World Allergy Organ. J. 2008, 1, 96–102.
- Adam, J.; Pichler, W.J.; Yerly, D. Delayed drug hypersensitivity: Models of T-cell stimulation. Br. J. Clin. Pharmacol. 2011, 71, 701–707.
- Schnyder, B.; Mauri-Hellweg, D.; Zanni, M.; Bettens, F.; Pichler, W.J. Direct, MHC-dependent presentation of the drug sulfamethoxazole to human alphabeta T cell clones. J. Clin. Investig. 1997, 100, 136–141.
- Kindmark, A.; Jawaid, A.; Harbron, C.G.; Barratt, B.J.; Bengtsson, O.F.; Andersson, T.B.; Carlsson, S.; Cederbrant, K.E.; Gibson, N.J.; Armstrong, M.; et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenom. J. 2008, 8, 186–195.
- Wei, C.Y.; Chung, W.H.; Huang, H.W.; Chen, Y.T.; Hung, S.I. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J. Allergy Clin. Immunol. 2012, 129, 1562–1569.e1565.
- Ostrov, D.A.; Grant, B.J.; Pompeu, Y.A.; Sidney, J.; Harndahl, M.; Southwood, S.; Oseroff, C.; Lu, S.; Jakoncic, J.; de Oliveira, C.A.; et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. USA 2012, 109, 9959–9964.
- Yun, J.; Cai, F.; Lee, F.J.; Pichler, W.J. T-cell-mediated drug hypersensitivity: Immune mechanisms and their clinical relevance. Asia Pac. Allergy 2016, 6, 77–89.
- Li, A.P. A review of the common properties of drugs with idiosyncratic hepatotoxicity and the “multiple determinant hypothesis” for the manifestation of idiosyncratic drug toxicity. Chem. Biol. Interact. 2002, 142, 7–23.
- Ulrich, R.G. Idiosyncratic toxicity: A convergence of risk factors. Annu. Rev. Med. 2007, 58, 17–34.
- Roth, R.A.; Maiuri, A.R.; Ganey, P.E. Idiosyncratic Drug-Induced Liver Injury: Is Drug-Cytokine Interaction the Linchpin? J. Pharmacol. Exp. Ther. 2017, 360, 461–470.
- Shaw, P.J.; Ganey, P.E.; Roth, R.A. Idiosyncratic drug-induced liver injury and the role of inflammatory stress with an emphasis on an animal model of trovafloxacin hepatotoxicity. Toxicol. Sci. 2010, 118, 7–18.
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36.
- Kato, R.; Uetrecht, J. Supernatant from Hepatocyte Cultures with Drugs That Cause Idiosyncratic Liver Injury Activates Macrophage Inflammasomes. Chem. Res. Toxicol. 2017, 30, 1327–1332.
- Mak, A.; Kato, R.; Weston, K.; Hayes, A.; Uetrecht, J. Editor’s Highlight: An Impaired Immune Tolerance Animal Model Distinguishes the Potential of Troglitazone/Pioglitazone and Tolcapone/Entacapone to Cause IDILI. Toxicol. Sci. 2018, 161, 412–420.