Brain arteriovenous malformations (BAVMs) are predominantly congenital vascular disorders that may arise anywhere inside of the central nervous system. They are comprised of one or more arterial feeders supplying a vascular nidus, and one or more draining veins. The nidus itself represents the site where arterial blood is shunted directly into the venous system without an interpolating network of capillaries.
- brain arteriovenous malformations
- cerebral cavernous malformations
- microRNA
1. Introduction
Brain arteriovenous malformations (BAVMs) are predominantly congenital vascular disorders that may arise anywhere inside of the central nervous system [1,2,3,4,5,6]. The nidus itself represents the site where arterial blood is shunted directly into the venous system without an interpolating network of capillaries. As a result, the blood flows at high pressure through these lesions, making them susceptible to hemorrhagic rupture, this being their most common form of presentation. A small number have also been described as appearing later in life after a different cerebral injury (trauma, infection, stroke, tumors, etc.), making their pathophysiology and genetic determination all the more intriguing and poorly understood [7,8].
Cerebral cavernous malformations (CCMs), or cavernomas, are vascular lesions that, unlike BAVMs, are generally small and well-delineated, with a low blood flow within them [9,10,11,12,13]. They are mostly asymptomatic and discovered incidentally on imaging studies; however, they can present via epilepsy or hemorrhagic stroke. The sporadic occurrence of CCMs typically denotes a single isolated lesion possibly linked to a developmental venous anomaly (DVA), whereas the familial variety leads to the formation of multiple lesions spread diffusely within the brain. Both BAVMs and CCMs can be effectively treated via microsurgical resection, ensuring symptomatic control and prevention of any future hemorrhage occurring from these lesions.
MicroRNAs (miRNA or miR) represent short strands of non-coding ribonucleic acid (RNA), approximately 20–22 nucleotides long, with the capacity to regulate the expression of target genes via the complete or partial binding onto their respective complementary sequences [17,18,19,20,21,22]. As a result, they hamper translation while also stimulating mRNA degradation. Currently, there is a shortage of data concerning the multitude of roles miRNAs play in the pathophysiology of AVMs and CCMs. In the following, we will present a summary of the miRNAs discovered to be dysregulated in these vascular pathologies, along with the pathways affected in other.
2. Methods
We explored the electronic databases of Medline (PubMed, PubMed Central) by employing various permutations of the terms and idioms: ‘arteriovenous malformation’, ‘AVM’, and ‘BAVM’, or ‘cavernous malformation’, ‘cavernoma’, and ‘cavernous angioma’ on the one hand, and ‘microRNA’, ‘miRNA’, and ‘miR’ on the other. Afterward, using cross-reference search, we then evaluated further articles pertaining to the individual miRNAs found in other cerebral diseases. We have considered both clinical studies on adult human patients and their pathological specimens, regardless of design, as well as experimental studies performed in vitro and in vivo. This non-systematic review may be significant in light of the increasing interest in the effective management and understating of cerebral vascular malformations.
3. microRNAs Involved in Cerebral Vasculogenesis and Angiogenesis
In the early embryonic life, the hemangioblasts emerge from the mesoderm as a response to local fibroblast growth factor (FGF) signaling. The surrounding endoderm secrets isoforms of vascular endothelial growth factor (VEGF) and Notably, while knockout of VEGFR2 is correlated with the lack of primitive vascular plexi [24], in Dicerex1/2 mice, even if the blood vessels are thin and disorganized, the presence of the vascular bedding suggests that miRNAs are not crucial for vasculogenesis, but for the next phase of vascular development: angiogenesis [25]. Indeed, Dicerex1/2 mice associated lower levels of Tie1 while VEGF, VEGFR1 and VEGFR2 were upregulated, but this is explained by the formation of abnormal blood vessels that favor local hypoxia and respond via VEGF activation accordingly.
After vasculogenesis, the endothelial cells (ECs) from the primary vascular plexus generate new capillaries by sprouting and non-sprouting angiogenesis. Sprouting angiogenesis is typically seen in brain formation and it is also the prototype for wound healing and tumor angiogenesis whereas non-sprouting angiogenesis involves splitting and fusion of capillaries and mainly occurs in lung development. In physiological sprouting angiogenesis, FGF and VEGF stimulation of the ECs induces the activation of matrix metalloproteinases (MMPs): MMP-2 and MMP-9, thus promoting the lysis of the basement membrane and the extracellular matrix (ECM) in the vicinity [26]. Not only FGF and VEGF are important in sprouting angiogenesis, but also angiopoietins (Ang1, Ang2, and Ang4) which bind to Tie1 and Tie2 receptors to modulate the response to VEGF.
Subsequently to the generation of the vascular network, remodeling, pruning and maturation phenomena occur in order to form the functional adult circulatory system. In adult life, angiogenesis is restricted to wound healing, inflammation, and reproduction. Therefore, in physiological conditions, the most abundant miRs in ECs are both pro-angiomiRs (miR-21, miR-126, let-7 family, miR-23, and miR-17-92 cluster) and anti-angiomiRs (miR-221, miR-222, and miR-24)
For instance, let-7 family members possibly target thrombospondin-1 (TSP-1), an endogenous inhibitor of angiogenesis. Dicer and Drosha knockout in human umbilical vein endothelial cells (HUVECs) results in the upregulation of TSP-1, probably by the decrease of let-7 family [34]. Another study revealed that Dicer blockade influences angiogenesis through miR-17-5p and let-7b which reduce tissue inhibitor of metalloproteinase 1 (TIMP1) expression and consequently MMPs activity [35]. The other two members of the miR-17-92 cluster, namely miR-18a and miR-19a, were also demonstrated to repress proteins containing thrombospondin type 1 repeats (TSR),
In hypoxic conditions, HIF-1α contributes to the alteration of miRNAs in HUVECs by upregulating miR-210, let-7a, let-7e, miR-103, miR-107, and to a lesser extent However, miR-328 is an anti-angiomiR whose target CD44 is involved in EC-EC and EC-ECM interactions [43]. Strikingly, let-7a, let-7e, miR-103, and miR-107 have AGO1 as a putative target and by the inhibition of AGO1-RISC complex, they produce a release in mRNAs that are suppressed during normoxia. Under VEGF stimulation, miR-155, miR-191, miR-21, and some of the miR-17-92 cluster (miR-18a, miR-17-5p, and miR-20a) are overexpressed in ECs, also shifting the balance towards angiogenic factors [44].
4. microRNAs in Cerebral Cavernous Malformations
Below, the miRNAs linked to CCMs are discussed, along with other illnesses possibly influenced by them. miR-27a, miR-125a, and mmu-miR-3472a have been individually studied in several other diseases, as well as in association with other similar acting microRNAs. disclosed 52 dysregulated miRNAs in brainstem CCM patients compared to controls, 10 of which were upregulated and 42 downregulated [119]. A more stringent statistical analysis narrowed down five miRNAs (let-7b-5p, miR-361-5p, miR-370-3p, miR-181a-2-3p, and miR-95-3p) as the most significant for CCM, which are discussed ulteriorly in this section, apart from the previously reviewed let-7b-5p.
, miR-27a was noticed to be increased in BECs isolated from in vivo mouse model brains harboring early CCM lesions and in cultured BECs displaying either CCM1 or CCM2 depletion [127]. On the other hand, the intraventricular delivery of a miR-27a-3p mimic in rats suffering from collagenase-induced intracerebral hemorrhage induced mitigation of brain edema, microvascular leakage, and leukocyte infiltration, as well as inhibition of neuronal apoptosis and microglial activation within the perihematomal zone by suppressing the overexpression of aquaporin-11 in BECs [131]. Similarly, the induced overexpression of miR-27a in rat brains before traumatic brain injury markedly diminished the neurological deficits and cerebral injury, particularly repressing autophagic activation after trauma occurred [132]. A comparable research performed on mice showed that the administration of mimics of miR-27a along with miR-23a considerably reduced the activation of proapoptotic Bcl-2 family constituents Bax, Noxa, and Puma lessened the severity of cortical lesion volume and hippocampal neural cell loss, and also undermined markers of caspase-dependent and -independent apoptosis after traumatic brain injury [133].
The downregulation of miR-125a has also been observed in children with epilepsy, despite the fact that no connection could be established between this miRNA and the inflammatory cytokines IFN-γ and TNF-α [137]. In rats, miR-125a was decreased in the blood of all models presenting seizures triggered by ischemia, hemorrhage, or kainite toxicity [138], as well as within the hippocampus after pentylenetetrazol-induced epilepsy [139]. Furthermore, the overexpression of miR-125a-5p resulted in a reduction of seizures and lessened hippocampal inflammation via the suppression of its target gene, calmodulin-dependent protein kinase IV (CAMK4) Pan et al. showed that microvesicles carrying miR-125a-5p fostered the proliferation, migration, the ability to form tubes of transfected microvessel BECs, improved their apoptotic rate [141], while an earlier study discovered the role of this microRNA in protecting BECs from the injurious effects of oxidized low-density lipoproteins such as senescence, a decrease of nitric oxide (NO), and upsurge of reactive oxygen species (ROS) [142].
More interestingly, they discovered that mmu-miR-3472a has cullin-associated and neddylation dissociated 2 (CAND2) as the putative target, which was often dysregulated in the transcriptomes of all three models researched (acute, chronic, and control). Also in this study, CAND2, which encodes TIP120b, a muscle-specific TATA-binding protein that is crucial in the process of myogenesis [143,144,145], was downregulated within in vitro BECs, though upregulated within the in vivo neurovascular units (NVUs) of acute and chronic models [143]. CAND2 is associated with the signaling pathway of cullins [146], also coordinating the activity of the Skp, Cullin, and F-box containing complex (SCF complex). Moreover, cullins are known to control the levels of VEGF2R on the surface of vascular endothelial cells via mRNA expression [147], as well as modulate the function of the endothelial barrier through the expression of vascular endothelial (VE)-cadherin, a key constituent of endothelial cell-to-cell adherens junctions.
This microRNA was observed as downregulated in the brainstem cavernomas included in the recent GWAS, with possible functional significance [119]. miR-361-5p has been established as a VEGF modulator in coronary artery disease [149], is implicated in the functionality of the blood-brain barrier including the formation of adherens junctions, lymphocyte adhesion to endothelial cells, and the integrity of the extracellular matrix [150], as well as an overexpressed serum biomarker in patients with acute myocardial infarction, ischemic stroke, and pulmonary embolism [151]. In patients with multiple sclerosis, its overexpression has also been correlated with decreased disease severity, suggesting a neuroprotective effect [150]. Because of these multiple involvements, miR-361-5p may represent a valid therapeutic target or biomarker for CCMs, although more research on its exact mechanisms is needed.
Lulli et al. have shown that miR-370-3p expression is markedly downregulated in gliomas when compared to normal brain tissue and neural stem cells [153]. The recovery of miR-370-3p expression in glioblastoma stem-like cells considerably reduced their proliferation, migration, and clonogenic abilities both in vitro, and in vivo models. This translates into a marked repressive effect on glioma cell proliferation by miR-370-3p overexpression or rescue. Another possible implication of miR-370-3p could be in primary progressive multiple sclerosis, wherein patients demonstrated significantly dysregulated serum levels of several microRNAs compared to controls, including this one [157].
Aside from the downregulation in CCM patients [119], low levels of miR-181a-2-3p, which targets FOXP2, have been linked to a poorer prognosis in patients with glioblastoma, although the exact acting mechanism of FOXP2 in these malignant tumors remains unclear [158]. miR-181a-2-3p is also downregulated under hypoxic conditions in glioblastoma cell lines in vitro [159] and has displayed a statistically significant differential expression between grade III and grade IV glioma cells, with an inferior level within the latter [160]. However, we could not find any additional information correlating this miRNA to other cerebrovascular malformations.
In an earlier research, it had been shown that miR-95-3p levels were positively correlated to glioma grade, and that a reduction in this microRNA elicited a similar response regarding the ability of glioma cells to proliferate and invade, but enhanced apoptosis [161]. Conversely, the overexpression of miR-95-3p caused a decrease in invasiveness, proliferation, and clonogenicity of metastatic lung adenocarcinoma within the brain by inhibiting oncogene cyclin D1 expression, and as a result prolonging metastasis-free survival [162]. Future studies may shed a light on whether miR-95-3p possesses a pathophysiological role in CCM development, or if its downregulation in this type of lesion was merely incidental. Table 1 summarizes the miRNAs discovered in CCMs, time of discovery, relative expression levels, and possible implications, and Figure 1 illustrates the current findings regarding their signaling pathways and putative targets.
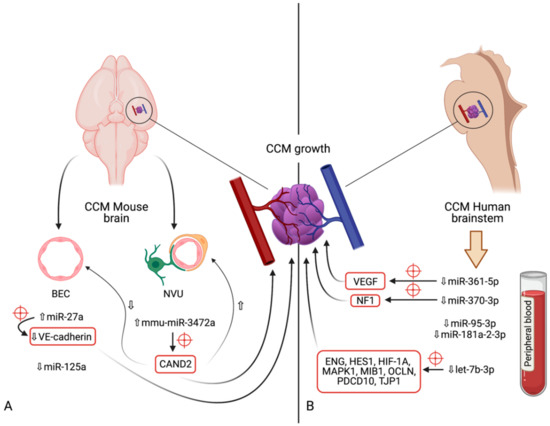
Figure 1. Diagram indicating the microRNAs and their respective expression levels, putative targets (marked by red crosshairs) and pathways involved in the development of cerebral cavernous malformations. The microRNAs are divided according to the model in which their expression was determined: (A)—mouse models (in vivo); (B)—human brainstem (according to the genome-wide association study by Kar et al. [97]). An upward pointing transparent arrow signifies upregulation; a downward pointing transparent arrow denotes downregulation. Abbreviations (in alphabetical order): BEC, brain endothelial cell; CAND2, cullin associated and neddylation dissociated 2; CCM, cerebral cavernous malformation; ENG, endoglin; HES1, Hes family bHLH transcription factor 1; HIF-1A; hypoxia inducible factor 1α; HMGA2, high-mobility group AT-hook 2; MAPK1, mitogen-activated protein kinase 1; MIB1, mindbomb E3 ubiquitin protein ligase 1; miR, microRNA; NVU, neurovascular unit; OCLN, occludin; PDCD10, programmed cell death 10; TJP1, tight junction protein 1; VE-cadherin, vascular endothelial cadherin.
Table 1. Location, Expression and Putative Targets of Identified microRNAs in Cerebral Cavernous Malformations.
| Author | miRNA | Location | Expression in CCM | Putative Targets | Known Cerebral Pathologies Associated (Expression)—Respective Putative Target(s) |
|---|---|---|---|---|---|
| Li et al., 2020 [105] | miR-27a | BEC (mouse model) | Upregulated | VE-cadherin | Temporal lobe epilepsy (upregulated)—GLRA2 Neuroprotection in rat models after TBI—Bax, Noxa, and Puma//FoxO3a |
| Li et al., 2020 [105] | miR-125a | Downregulated | NS | Epilepsy—CAMK4 Ischemic stroke |
|
| Koskimäki et al., 2019 [121] | mmu-miR-3472a | Mouse NVU | Dysregulated | CAND2 | NS |
| Kar et al., 2017 [97] | miR-361-5p | Brainstem CCM | Downregulated | VEGF, EGFL7 | Acute myocardial infarction Ischemic stroke Pulmonary embolism Multiple sclerosis Intellectual disability |
| Kar et al., 2017 [97] | miR-370-3p | Downregulated | NF1 | Glioma (downregulated)—HMGA2, HIF-1A, NEAT1//CTBP1-AS2 Sepsis associated encepahlopathy in mouse models |
|
| Kar et al., 2017 [97] | miR-181a-2-3p | Downregulated | NS | Glioblastoma (downregulated)—FOXP2 | |
| Kar et al., 2017 [97] | miR-95-3p | Downregulated | NS | Glioma (upregulated)—CELF2 Lung cell adenocarcinoma metastasis (downregulated)—cyclin D1 |
|
| Kar et al., 2017 [97] | let-7b-5p | Downregulated | ENG, HES1, HIF-1A, MAPK1, MIB1, OCLN, PDCD10, TJP1 |
BAVM (upregulated) Epilepsy (downregulated) Ischemic stroke models (downregulated) Glioma (downregulated)—IKBKE, E2F2, oncogenes KRAS, HMGA2 and MYC |
Abbreviations (in alphabetical order): BAVM, brain arteriovenous malformation; BEC, brain endothelial cell; CAMK4, calmodulin-dependent protein kinase IV; CAND2, cullin associated and neddylation dissociated 2; CCM, cerebral cavernous malformation; CELF2, CUGBP- and ETR-3-like family 2; EGFL7, epidermal growth factor like domain 7; ENG, endoglin; FoxO3a, Forkhead box O3a; GLRA2, glycine receptor alpha 2; HES1, Hes family bHLH transcription factor 1; HIF-1A; hypoxia inducible factor 1α; HMGA2, high-mobility group AT-hook 2; KLF2, kruppel-like factor 2; MAPK1, mitogen-activated protein kinase 1; MIB1, mindbomb E3 ubiquitin protein ligase 1; miRNA and miR, microRNA; NEAT1, Nuclear Enriched Abundant Transcript 1; NS, not specified; NVU, neurovascular unit; OCLN, occludin; PDCD10, programmed cell death 10; TBI, traumatic brain injury; TJP1, tight junction protein 1; VE-cadherin, vascular endothelial cadherin.
This entry is adapted from the peer-reviewed paper 10.3390/cells10061373