Cardiovascular disease is the leading cause of morbidity and mortality in the western and developing world, and the incidence of cardiovascular disease is increasing with the longer lifespan afforded by our modern lifestyle. Vascular diseases including coronary heart disease, high blood pressure, and stroke comprise the majority of cardiovascular disease and therefore represent a significant medical and socioeconomic burden on our society. It is not be surprising that these conditions overlap and potentiate each other when we consider the many cellular and molecular similarities between them. At the molecular level, the vascular smooth muscle cell (VSMC) is the target, integrator, and effector cell of both atherogenic and the major effector protein of the hypertensive signal, Angiotensin II (Ang II). Together, these signals can potentiate each other and prime the artery and exacerbate hypertension and atherosclerosis. Therefore, VSMC are the fulcrum in progression of these diseases and therefore, understanding the effects of atherogenic stimuli and Ang II on VSMC is key to understanding and treating atherosclerosis and hypertension. In this review, we will examine studies in which hypertension and atherosclerosis intersect on the VSMC, and illustrate common pathways between these two diseases and vascular aging.
- vascular smooth muscle cell
- Angiotensin II
- hypercholesterolemia
- atherosclerosis
- hypertension
- vascular disease
1. Definition
Vascular diseases including coronary heart disease, high blood pressure, and stroke comprise the majority of cardiovascular diseases, and therefore represent a significant medical and socioeconomic burden on our society. It may not be surprising that these conditions overlap and potentiate each other when we consider the many cellular and molecular similarities between them. These intersecting points are manifested in clinical studies in which lipid lowering therapies reduce blood pressure, and anti-hypertensive medications reduce atherosclerotic plaque. At the molecular level, the vascular smooth muscle cell (VSMC) is the target, integrator, and effector cell of both atherogenic and the major effector protein of the hypertensive signal Angiotensin II (Ang II). Together, these signals can potentiate each other and prime the artery and exacerbate hypertension and atherosclerosis. Therefore, VSMCs are the fulcrum in progression of these diseases and, therefore, understanding the effects of atherogenic stimuli and Ang II on the VSMC is key to understanding and treating atherosclerosis and hypertension.
2. Introduction
2.1. Hypertension and Angiotensin II
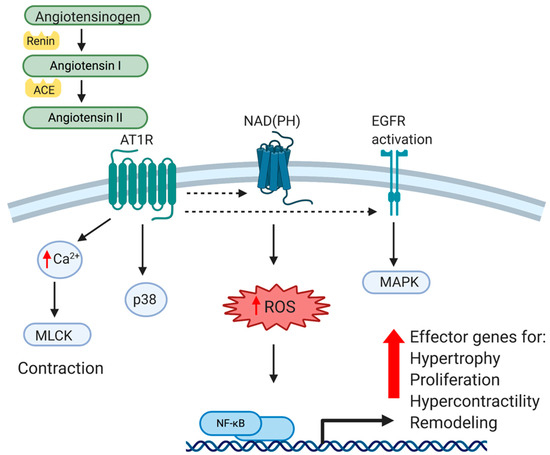
3. Data, Model, Applications and Influences
3.1. Angiotensin II, Atherosclerosis, and Vascular Aging
3.1.1. Aging and Angiotensin II
3.1.2. Aging and Atherosclerosis
4. Conclusions
The synthesis of the studies outlined in this review present an intimate relationship between hypertension, atherosclerosis, and aging. Chronic hypercholesterolemia induces the expression of many components of the RAS, and at multiple systemic and cellular targets, Ang II induces the expression of inflammatory and atherogenic proteins, which together leave the artery primed for hypertension and atherosclerosis. Inhibitors of Ang II synthesis and AT1R antagonists reduce vascular inflammation and plaque progression, and lipid-lowering therapy reduces blood pressure. In many murine studies, the genetic deletion of multiple components of the atherosclerotic pathway reduces hypertension, and deletion of members of the RAS attenuates atherogenesis. These studies are supported by clinical observations in humans using pharmacological inhibitors. Both of these diseases are more prevent in aged arteries, and their severity is exacerbated by age. Morphologically, advanced hypertensive and atherosclerotic arteries resemble those from aged individuals. Atherogenic and hypertensive stimuli intersect on the VSMC suggesting that hypertension, atherosclerosis, and vascular smooth muscle cells make a perfect trio for vascular pathophysiology. The identification of the molecular mediators of these diseases is key to our better understanding of them and, when supported by rigorous genetic studies in mice, represents an opportunity for the development of therapeutics to combat these vascular diseases which are approaching epic proportions.
References
- Wang, M.; Monticone, R.E.; Lakatta, E.G. Proinflammation of aging central arteries: A mini-review. Gerontology 2014, 60, 519–529. [Google Scholar] [CrossRef]
- Asai, K.; Kudej, R.K.; Shen, Y.T.; Yang, G.P.; Takagi, G.; Kudej, A.B.; Geng, Y.J.; Sato, N.; Nazareno, J.B.; Vatner, D.E.; et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1493–1499. [Google Scholar] [CrossRef]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Gepner, A.D.; Korcarz, C.E.; Colangelo, L.A.; Hom, E.K.; Tattersall, M.C.; Astor, B.C.; Kaufman, J.D.; Liu, K.; Stein, J.H. Longitudinal effects of a decade of aging on carotid artery stiffness: The multiethnic study of atherosclerosis. Stroke 2014, 45, 48–53. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Spinetti, G.; Jiang, L.Q.; Monticone, R.; Zhao, D.; Cheng, L.; Krawczyk, M.; Talan, M.; Pintus, G.; et al. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am. J. Pathol. 2005, 167, 1429–1442. [Google Scholar] [CrossRef]
- Bruno, R.M.; Duranti, E.; Ippolito, C.; Segnani, C.; Bernardini, N.; Di Candio, G.; Chiarugi, M.; Taddei, S.; Virdis, A. Different Impact of Essential Hypertension on Structural and Functional Age-Related Vascular Changes. Hypertension 2017, 69, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Iwanami, J.; Jing, F.; Tsukuda, K.; Ohshima, K.; Horiuchi, M. Angiotensin II type 2 receptor-interacting protein prevents vascular senescence. J. Am. Soc. Hypertens. 2012, 6, 179–184. [Google Scholar] [CrossRef]
- Shan, H.Y.; Bai, X.J.; Chen, X.M. Apoptosis is involved in the senescence of endothelial cells induced by angiotensin II. Cell Biol. Int. 2008, 32, 264–270. [Google Scholar] [CrossRef]
- Weiss, D.; Sorescu, D.; Taylor, W.R. Angiotensin II and atherosclerosis. Am. J. Cardiol. 2001, 87, 25–32. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Monticone, R.E.; Lakatta, E.G. Proinflammation of aging central arteries: A mini-review. Gerontology 2014, 60, 519–529. [Google Scholar] [CrossRef]
- Asai, K.; Kudej, R.K.; Shen, Y.T.; Yang, G.P.; Takagi, G.; Kudej, A.B.; Geng, Y.J.; Sato, N.; Nazareno, J.B.; Vatner, D.E.; et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1493–1499. [Google Scholar] [CrossRef]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Gepner, A.D.; Korcarz, C.E.; Colangelo, L.A.; Hom, E.K.; Tattersall, M.C.; Astor, B.C.; Kaufman, J.D.; Liu, K.; Stein, J.H. Longitudinal effects of a decade of aging on carotid artery stiffness: The multiethnic study of atherosclerosis. Stroke 2014, 45, 48–53. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Spinetti, G.; Jiang, L.Q.; Monticone, R.; Zhao, D.; Cheng, L.; Krawczyk, M.; Talan, M.; Pintus, G.; et al. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am. J. Pathol. 2005, 167, 1429–1442. [Google Scholar] [CrossRef]
- Bruno, R.M.; Duranti, E.; Ippolito, C.; Segnani, C.; Bernardini, N.; Di Candio, G.; Chiarugi, M.; Taddei, S.; Virdis, A. Different Impact of Essential Hypertension on Structural and Functional Age-Related Vascular Changes. Hypertension 2017, 69, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Iwanami, J.; Jing, F.; Tsukuda, K.; Ohshima, K.; Horiuchi, M. Angiotensin II type 2 receptor-interacting protein prevents vascular senescence. J. Am. Soc. Hypertens. 2012, 6, 179–184. [Google Scholar] [CrossRef]
- Shan, H.Y.; Bai, X.J.; Chen, X.M. Apoptosis is involved in the senescence of endothelial cells induced by angiotensin II. Cell Biol. Int. 2008, 32, 264–270. [Google Scholar] [CrossRef]
- Weiss, D.; Sorescu, D.; Taylor, W.R. Angiotensin II and atherosclerosis. Am. J. Cardiol. 2001, 87, 25–32. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
This entry is adapted from the peer-reviewed paper 10.3390/ijms21124525