Long noncoding RNAs exceeding a length of 200 nucleotides play an important role in ensuring cell functions and proper organism development by interacting with cellular compounds such as miRNA, mRNA, DNA and proteins. However, there is an additional level of lncRNA regulation, called lncRNA epigenetics, in gene expression control.
- lncRNA epigenetics
- RNA modyfiing enzymes
- detection modified nucleotides
1. General Remarks
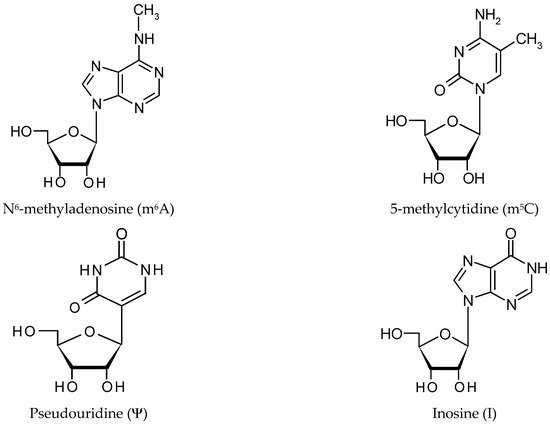
2. RNA Modification Mechanisms
There are three groups of protein involved in modifying RNA metabolism [27,28]. The first group (writers,Figure 2) consists of enzymes introducing modified nucleotides into RNA during posttranscriptional RNA modifications; the second group of proteins interacts with modified nucleotides (readers); and the third group is involved in removing modification labels (erasers).
In humans, the formation of the m6A modification is connected with the methylase complex (writer). Crystallographic and biochemical studies have shown that METTL3 is S-adenosylmethionine methyltransferase with catalytic properties, while METTL14 serves as an RNA binding platform (Figure 2A) [48]. Recently, METTL16 was characterized as another “writer” protein, which interacts with several types of RNAs: mRNA, U6 and lncRNA [58,59,60,61]. Unlike METTL3/METTL14, which modifies A to m6A in coding RNAs, METTL16 can methylate both coding and noncoding RNAs [62].
Like DNA and histone modifications pathways, the m6A modification has two specific erasers, FTO and ALKBH5 [64,65]. FTO (fat mass and obesity-associated protein) removes the methylation trace by oxidizing m6A to N6-hydroxymethyladosine or N6-formyladenosine, which are chemically unstable and can hydrolyze to the final adenine product [66,67,68]. Another eraser is the homologous protein ALKBH5, which catalyzes the direct removal of the methyl group from adenine [69]. Whereas the FTO protein has been linked to obesity, the ALKBH5 protein is essential to spermatogenesis.
The human YTH domain containing protein family consists of five proteins, namely, YTHDF1–3 (eIF3) binds to the m6A located in the 5′UTR region of mRNA and is involved in cap-independent translation [82,83,84]. This mechanism is known as the “RNA epigenetic m6A switch”, which means m6A alters the local structure of mRNA or lncRNA, to facilitate the binding of HNRNPs for biological regulation [87].
Two writer m5C methyltransferases (MTases) have been shown to catalyze the m5C modification of eukaryotic RNA. One of them, RNA DNMT2, resembles DNA methyltransferases in its structure and characteristics [92,93], whereas the second group comprised of seven members, the MTPases (NSUN), contains the conserved NOL1/Nop2/Sun motif [94]. In 2017, Yang et al. presented evidence through in vitro and in vivo studies that m5C formation in mRNAs is mainly catalyzed by the NSUN2 type RNA methyltransferase [95]. Moreover, hm5C modifications of RNA are involved in stem cell pluripotency and impact translation efficiency [97,98].
[28]. Ψ has an unusual nucleoside, containing a C–C glycosidic bond, instead of the N-C bond found in the rest of the nucleosides [100]. As a result of U being replaced by Ψ, an additional hydrogen bond donor is present at the non-Watson–Crick edge. The distinct structure of Ψ increases both the rigidity of the phosphodiester backbone, as well as the thermodynamic stability of Ψ–A, compared with U–A [101,102].
Pseudouridine writers, called pseudouridine synthases (PUSs), recognize substrates and catalyze the isomerization of U to Ψ, without the need for cofactors (Figure 2B) [42,100,103]. However, PUS enzymes are unable to isomerize free nucleotides. In rRNA pseudouridylation, small nucleolar RNAs act as guides that recognize targets with sequence complementarity, thus directing pseudouridylation in a site-specific manner [104,105,106]. In the solved crystal structure, three of the proteins interact with the H/ACA guide RNA or substrate RNA, while GAR1 may regulate substrate loading and release [104,108].
In contrast to the m6A modification, specific Ψ eraser or reader proteins have not yet been identified. Furthermore, the lack of reader proteins that specifically bind to Ψ means it is difficult for proteins to identify the more subtle modification, which results in the C–N bond being replaced by a C–C bond, between uracil and ribose [109]. In humans, 10 proteins (PUS1–10) involved in RNA modification, with an annotated Ψ synthase domain, have been found (writers). It is also possible that there could be readers and erasers for the Ψ modification; however, their existence has not yet been proven.
ncRNAs and is catalyzed by the writer protein, adenosine deaminase acting on RNA (ADAR). ADAR1 and ADAR2 catalyze all the currently known A-to-I editing sites. Inosine essentially mimics the chemical properties of guanosine, therefore ADAR proteins introduce an A-to-G substitution in transcripts. These changes can lead to specific amino acid substitutions, altering protein composition.
3. Detection of Modified Nucleotides
The information presented above suggests that lncRNA epigenetics are important to cell differentiation and may be involved in controlling organism development. This has motivated many laboratories to screen ncRNAs for the presence of modified nucleotides, and to study the changes of the modification pattern during cell development [26,27].
Application NGS methods to epigenetic mapping is so far difficult because they typically do not detect modified nucleosides [116,117]. Developing single-base resolution sequencing, which could quantify the relatively low abundance of modified nucleotides in lncRNA, is a significant challenge. The identification of transcriptome-wide RNA modifications has been approached using different strategies.
The study of RNA modification started in 1957 when the first modified nucleoside, pseudouridine, was discovered in bulk yeast RNA, using paper chromatography [118]. The cut site is labeled with32P and the32P labeled RNA fragment is splint ligated to 116-nucleotide single stranded DNA oligonucleotide, using DNA ligase. The sample is then digested with RNase T1/A to completely digest all RNA, whereas the32Plabelled candidate site remains with the DNA nucleotide as DNA-32P(A/m6A)p and DNA-32P(A/m6A)Cp, which migrate as 117/116 mers on denaturing gel. This method was successfully used to determine the modified nucleotides like m6A, m5C, Ψ, and possibly other unknown modified nucleotides, in several coding and noncoding RNAs.
The high-throughput m6A mapping strategies were based on the immunoprecipitation of modified RNA molecules, using m6A-specific antibodies coupled to the subsequent NGS sequencing. Then, the RNA separated with a magnet is subjected to a second round of m6A immunoprecipitation. The resulting RNA pool, which is highly enriched with m6A-containing RNAs, is used for library construction and NGS sequencing [117,126]. miCLIP allows for a high-resolution detection of m6A in RNAs.
The detection of the m6A nucleotide in RNA, using an indirect approach, is difficult because few chemical reagents modify the methyl group. However, NOseq, a method for the detection of m6A in RNA after chemical deamination by nitrous acid, has recently been introduced [130]. Nitrous acid deaminates adenosines to inosine, while the m6A residue is resistant to such modifications. The application of NGS after modification to detect m6A sites in MALAT1 lncRNA
The most common indirect method used to determine m5C modification sites in RNA, i.e., the bisulfite conversion of cytidine to uridine has been applied successfully to determine the presence of this modified nucleotide in DNA [131]. The method is based on the fact that m5C modified cytosine is resistant to bisulfite cytosine deamination. There are several biochemical kits on the market, making it possible to prepare m5C libraries, which are ready for sequencing. Moreover, other modifications or double-stranded regions may be resistant to bisulfite treatment, especially under the milder reaction conditions required to maintain RNA integrity.
An alternative approach, which has been termed the “suicide enzyme trap”, has been employed to characterize the substrates of the following m5C-methyltransferases (m5C-MTases), NSUN2 and NSUN4 [137,138]. By mutating m5C-MTases to form irreversible covalent bonds with target residues, the resulting stable enzyme–RNA complexes are suitable for immunoprecipitation and mapping. This is also the case with the AZA-seq methodology, in which the “suicide inhibitor” nucleotide analog of 5-azacytidine is incorporated into cellular RNA and “traps” m5C-MTases for pulldown and sequencing [139].
Determining the Ψ sites in the RNA chain requires indirectly analyzing this modification using carbodiimide chemistry. The bulky CMCT group attached to N3 on Ψ hinders reverse transcription and results in cDNA being truncated. This facilitates the detection of Ψ at a single nucleotide resolution level [140]. Sites of pseudouridylation with single nucleotide resolution can be identified by subjecting the data obtained through NGS sequencing of Pseudo-seq libraries, to computational analysis.
Recently, Pan et al. developed a method that uses a CMC-Ψ-induced RT stop with an additional step of site-specific ligation, followed by PCR, to generate two unique PCR products, that correspond to the modified and unmodified uridine. The modification is visualized in the PCR products using gel electrophoresis [143].
As identifying true editing sites from transcriptome sequencing data is difficult, alternative methods aimed at marking inosine have been developed. RNase T1specifically cleaves RNA after guanosine or inosine but is inhibited by guanosine glyoxal/borate adducts. The cleavage of glyoxal-modified RNA creates RNA fragments that carry inosine at their termini, as an input for sequencing. ICE involves the treatment of RNA with acrylonitrile, which converts the inosine to N1-cyanoethylinosine in the process of cyanoethylation, and results in the formation of an inosine/acrylonitrile adduct that inhibits base pairing with cytidine and stalls reverse transcription.
Recently, a direct modification detection method, called nanopore sequencing, has been developed [155,156,157]. Tombo is a software used to detect modifications in DNA and RNA, such as the m5C modification in DNA and RNA and the m6A modification in DNA [160]. The EpiNano software is used to detect the m6A modification in RNA [117,155]. It needs to be highlighted that direct RNA modification analysis using nanopore sequencing is rapidly developing and becoming more reliable, so its routine application in the field of RNA epigenetics is expected.
4. The Impact of lncRNA Epigenetics on lncRNA Function
It is estimated that human cells have over 50,000 lncRNA molecules coded in genes. Many mature lncRNAs are modified after transcription (Section 2). The application of NGS methods in combination with bioinformatic analysis revealed the occurrence of several modifications in different types of lncRNA molecules. recruit factors, either to the site of lncRNA transcription or to adjacent loci and involve lncRNA XIST and lncRNA H19 [165,166].
Due to its size, XIST has many modifications like 78 sites m6A. 5 sites m5C and single site of Ψ [26]. In humans, multiple m6A sites in XIST repeat regions have been identified. mRNAs, is mediated by RBM15 and RBM15B, which bind the m6A-methylation complex and recruit it to specific sites in RNA [83]. Additionally, the knockdown of RBM15 and RBM15B, or the knockdown of METTL3 methyltransferase, impairs XIST-mediated gene silencing.
Many pathways contribute to the control of gene expression during development. Polycomb repressive complex (PRC2) and XIST are associated with gene repression in various developmental processes, such as X chromosome inactivation and genomic imprinting.
PRC2 binds with high affinity to the 5′-end region of XIST called the repeat In human XIST, five m5C marks were also detected. The presence of m5C methylation in the XIST transcript prevents the binding of the PRC2 in vitro. In Xist lncRNA, the presence of pseudouridylation and A–I editing sites has been confirmed, however, their roles are unknown at this stage so far [170,171].
The recent analysis of MeRIP seq data revealed thousands of m6A switches, which are involved in alternative RNA splicing and abundance [173]. They also show that the recognition of the m6A mark in MALAT1 by the YTHDC1 reader protein plays a critical role in maintaining the composition of nuclear speckles and their genomic binding sites. In addition,MALAT1lncRNA is subject to post-transcriptional m5C modification; five m5C sites been found to regulate chromatin-related roles in other lncRNAs, such as HOTAIR and XIST [135]. Although the exact role played by pseudouridine and inosine modifications remains to be explained, and the presence of each of the three inosines increases the stability of MALAT1 by 2–3 kcal/mol [28,170].
HOTAIR interacts with the nuclear m6A reader YTHDC1 at the methylated A783 and at additional sites [176]. Localization in chromatin strongly depends on the m6A modification at site A783 of HOTAIR, while the modification of other m6A sites mediates high HOTAIR levels. The previous results demonstrate that site-specific cytosine methylation occurs in lncRNA HOTAIR [135]. The methylation of C1683 is widespread in different cell types and it is not limited by the abundance of HOTAIR RNA levels.
This lincRNA is necessary for the differentiation of mouse embryonic stem cells (mESCs) and acts as a ceRNA by sequestering let-7 miRNAs [177]. It has been proposed that the presence of m6A in lincRNA1281 can act as a m6A-switch for specific RNA binding proteins, which will eventually regulate their interaction with let-7 miRNA. However, the identity of such proteins has not yet been discovered. A similar mechanism has already been proposed for the binding of the HuR (ELAVL1) protein and miRNAs, to the mRNAs encoding developmental regulators in mESCs.
H19, an imprinted lncRNA with a size of 2.3 kb, plays an important role in embryonic development [178]. The knockdown of METTL3 or METTL14 notably reversed the hypoxic preconditioning-induced (HPC-induced) enhancement of cell viability, anti-apoptosis ability, and lncRN AH19 expression [179]. The Ras-GTPase-activating protein-binding protein 1 (G3BP1) was confirmed to bind methylated lncRNAH19, based on the presence of NSUN2. lncRNA H19 has two editing sites, and their presence increases the lncRNA stabilization energy by 3 kcal/mol [171].
The human steroid receptor RNA activator (SRA) is a transcript of thesra1gene, whose size ranges from 0.7 to 0.9 kb. It is a dual-function RNA, which acts as both an lncRNA and an mRNA [181]. lncRNA SRA regulates several processes, such as cell cycle proliferation, as well as insulin, Notch, and TNFa signaling [182]. In a subsequent study, the same authors identified a specific uridine residue in SRA1 (U206), whose modification by PUS1 (or PUS3) might induce a functional switch, which regulates nuclear receptor signaling [26,181].
It serves an oncogenic role in a variety of malignant tumors, such as colorectal cancer [183]. PVT1 lncRNA is highly modified and contains m6A, m5C and Ψ Moreover, the RNA m6A modification mediated by the METTL3/METTL14 complex (Figure 2A) regulates epidermal stemness by controlling Pvt1 and MYC interactions through Pvt1 methylation, uncovering a key and novel molecular mechanism underlying skin tissue homeostasis, regeneration and wound repair. Some of the Ψ sites were located within functional lncRNA motifs, indicating the potential regulatory impact of Ψ on lncRNAs.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22116166