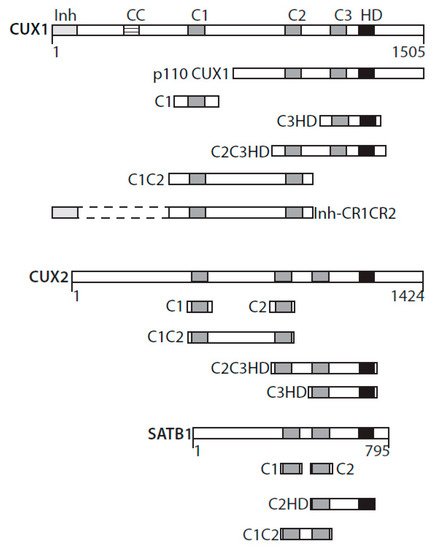
A role for CUX1 in DNA repair was first suggested from the analysis of mammary tumors that develop in MMTV-CUX1 transgenic mice [
9]. Accordingly, 44% of tumors from these transgenic mice exhibited a spontaneous missense mutation at codon 12 or 61 of
Kras that resulted in the activation of this oncogene [
9]. The prevalence of these mutations indicates that spontaneous activating mutations within
Kras occur frequently; however, we rarely observe tumors arising from such mutations in the wild-type mouse. This suggests that other cooperating events are needed for tumor development following activation of a
RAS gene. The cooperation between CUX1 and Kras was confirmed using lentiviral infections in the lung [
9]. Mice that received a CUX1-expressing lentivirus in addition to the Kras lentivirus developed a higher number of tumors than mice infected only with the Kras lentivirus [
9]. These tumors were of larger size and progressed further along the pathological spectrum. While the
KRASG12D mice developed solely grade 1 adenomas, mice expressing KRAS
G12D and CUX1 developed higher-grade adenomas and one large adenocarcinoma [
9]. That ectopic expression of KRAS
G12D alone produced only low-grade adenomas was not unexpected. Earlier studies showed that
RAS oncogenes cannot transform primary cells [
18]. Cells that harbor an activated RAS oncogene produce an excess of reactive oxygen species (ROS) that cause oxidative DNA damage and, ultimately, cellular senescence () [
19,
20,
21]. Cellular senescence can also be observed in cells that exhibit constitutive activation of the RAS pathway as a consequence of a mutation in another gene of the same signaling pathway (reviewed in [
22,
23]). This has been documented not only in tissue culture and mouse models [
24,
25,
26], but also in premalignant human colon adenomas [
27,
28,
29], as well as in human benign lesions caused by the
BRAFV600E mutation [
30] or NF1 inactivation [
31]. In this context, cellular senescence has been deemed a tumor suppression mechanism [
32]. Unfortunately, cancer cells can sometimes adapt and continue to proliferate despite producing high levels of ROS. Cancer cells can reduce ROS levels by increasing the expression of antioxidants [
33,
34,
35], notably following inactivation of the
KEAP1 tumor suppressor gene, an event observed in 15–30% of cancers [
36]. Alternatively, what is becoming increasingly evident is that cancer cells can adapt to elevated ROS by increasing their capacity to repair oxidative DNA damage [
9,
37,
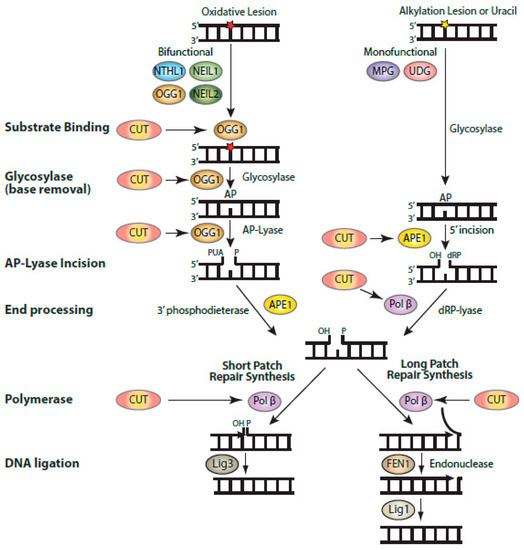
38]. This can be achieved through increased expression of enzymes of the base excision repair (BER) pathway, as well as BER accessory factors [
38]. Indeed, ectopic expression of CUX1 together with an activated RAS oncogene did not affect ROS levels, but greatly reduced genomic DNA damage, as well as the proportion of senescent cells in the population [
9]. Further experiments showed that CUX1 accelerated the repair of oxidative DNA damage caused by exposure to H
2O
2 [
9,
39]. In particular, single-cell gel electrophoresis (comet assay) performed at pH 10 after treatment with the formamidopyrimidine DNA glycosylase (FPG) enzyme revealed that CUX1 accelerates the repair of oxidized purines [
9,
39]. In turn, CUX1 knockdown delays the repair of oxidized purines [
9]. These results were confirmed by measuring the levels of 8-oxodeoxyguanines in genomic DNA [
9].
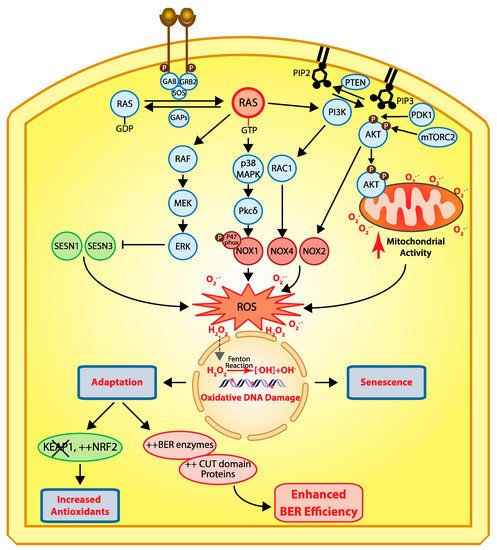
Figure 2. Cellular responses to RAS-driven production of reactive oxygen species. In physiological conditions, RAS proteins (KRAS, HRAS, and NRAS) alternate between their GDP- and GTP-bound states which are regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) [
40]. Binding of a receptor tyrosine kinase (RTK) to its ligand induces its dimerization and autophosphorylation, which in turn allows the recruitment of SH2 domain-containing proteins such as GRB2 that recruits the GEF protein SOS which then activates RAS [
41,
42]. In cancer, RAS is activated by a number of mechanisms including activating mutations in one of the RAS genes, overexpression of RTKs, and loss of GAP proteins (reviewed in [
43]). In its GTP-bound state, RAS activates a number of downstream effector pathways that elevate the level of reactive oxygen species (ROS) in multiple ways [
23]. The mitogen-activated protein kinase (MAPK) pathway leads to the transcriptional activation of NOX1 which codes for NADPH oxidase 1, a member of the NADPH oxidase enzyme family that catalyzes the one-electron transfer of oxygen to generate superoxide at the plasma membrane [
21,
44]. In addition, phosphorylation of the p47phox Nox1 subunit by protein kinase C δ (PKCδ) induces its translocation to the plasma membrane [
45]. Another study provided evidence that RAS-induced ROS production is dependent on RAC1 and NOX4, another member of the NADPH oxidase enzyme family [
46]. The RAF–MEK–ERK pathway causes the transcriptional repression of sestrin family genes, SESN1 and SESN3, which code for antioxidant modulators of peroxiredoxins [
47]. Another important signaling pathway downstream of RAS is the pleiotropic PI3K/AKT pathway [
43]. Importantly, the PI3K/AKT pathway is frequently activated in human cancers in a RAS-independent manner following a mutation or amplification of PIK3CA which codes for the p110α catalytic subunit of the phosphatidylinositol 3-kinase (PI3K) or following inactivation of PTEN, a gene encoding the tumor suppressor phosphatase and tensing homolog, which dephosphorylates phosphatidylinositol 3,4,5-triphosphate (PIP
3) to phosphatidylinositol 4,5-bisphosphate (PIP
2), thereby terminating PI3K-dependent signaling [
43]. The accumulation of PIP3 facilitates the localization of PH domain-containing proteins such as AKT and PDK1 to the plasma membrane where AKT is activated following its phosphorylation by PDK1 [
48,
49,
50] and mTORC2 [
51,
52,
53]. Activated AKT phosphorylates a large number of proteins involved in diverse cellular processes [
43]. Importantly, PI3K/AKT signaling has been implicated in the activation of NOX activity [
54,
55]. Moreover, phosphorylation of AKT induces its translocation to the mitochondrial matrix and inner membrane [
56], where it can phosphorylate GSK-3β, thereby lifting the negative regulation of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, two complexes that produce superoxide and H
2O
2 [
57,
58,
59]. Note that reactive oxygen species (ROS) do not travel through the cell since they react with the next molecule. However, ROS can be converted into H
2O
2 which moves into the cells and penetrates the nucleus, where it can be converted into hydroxyl radicals (
•OH
−) when it comes in contact with ferrous ions [
60]. Hydroxyl radicals in turn cause oxidative DNA damage, and sustained DNA damage eventually causes cellular senescence. Cells can adapt via two mechanisms: (1) increased expression of antioxidants following inactivation of KEAP1 and/or upregulation of NRF2 [
35], or (2) increased expression of BER enzymes and accessory factors such as CUT domains [
9,
10,
11,
38,
61,
62].
Subsequent experiments established that
CUX1 knockdown is synthetic lethal in all cancer cells exhibiting high levels of ROS due to an activating mutation in a
RAS gene (Hs578T
HRAS, MDA-MB-231
KRAS, DLD-1
KRAS, HCT116
KRAS, KE37
NRAS), another gene in the pathway (HT29
BRAF), or an upstream receptor tyrosine kinase (HCC827
EGFR) ([
9,
38] and Ramdzan et al., in preparation). In contrast, CUX1 knockdown did not reduce the clonogenic efficiency of cell lines that exhibit relatively low ROS levels [
38]. The case of the A549 cells is particularly enlightening. These cells carry an activating mutation in the
KRAS oncogene but still exhibit low ROS, because inactivation of the
KEAP1 tumor suppressor gene in these cells leads to greater accumulation of NRF2 in the nucleus and increased activation of genes coding for antioxidants [
36].