Lipopolysaccharides (LPSs) are bacterial surface glycolipids, produced by Gram-negative bacteria. LPS is known to determine acute inflammatory reactions, particularly in the context of sepsis. However, LPS can also trigger chronic inflammation. In this case, the source of LPS is not an external infection, but rather an increase in endogenous production, which is usually sustained by gut microbiota (GM), and LPS contained in food. The first site in which LPS can exert its inflammatory action is the gut: both GM and gut-associated lymphoid tissue (GALT) are influenced by LPS and shift towards an inflammatory pattern. The changes in GM and GALT induced by LPS are quite similar to the ones seen in IBD: GM loses diversity, while GALT T regulatory (Tregs) lymphocytes are reduced in number, with an increase in Th17 and Th1 lymphocytes. Additionally, the innate immune system is triggered, through the activation of toll-like receptor (TLR)-4, while the epithelium is directly damaged, further triggering inflammation.
1. Introduction
Lipopolysaccharides (LPSs) are bacterial surface glycolipids, produced by Gram-negative bacteria. It is present in the outer membrane of most Gram-negative bacteria, which is also made up of phospholipids. LPS is composed of three domains: lipid A, the core oligosaccharide, and the O antigen. Depending on the bacteria there can be differences, however this structure is usually maintained. The main function of LPS is structural, acting as a barrier against toxic agents for the bacteria, for instance, antibiotics. It also plays an important role in stimulating the immune system, given it is one of the first bacterial components the immune system encounters [
1].
Indeed, the role of LPS in stimulating inflammation has been acknowledged since the early 20th century. Pfeiffer was the first to be able to identify it and demonstrate it could cause septic shock, even though its structure was only described later. LPS can determine a very strong inflammatory reaction, mediated by its interaction with the innate compartment of the immune system [
2]. Toll-like receptor (TLR)-4 is the key interlocutor of LPS, determining cytokine cascades and caspase activation [
3].
LPS can determine a variety of complications other than septic shock and endotoxemia, particularly when it is chronically present at low levels. This condition is associated with metabolic endotoxemia, a condition in which alterations in the gut epithelial barrier allow microbiota-produced LPS to enter the bloodstream. Low-grade inflammation has been linked with many different diseases, such as diabetes, obesity, non-alcoholic fatty liver diseases, chronic kidney disease, and cardiovascular disease [
4]. It has also been linked to immune disruption in HIV [
5]. Not surprisingly, metabolic endotoxemia is typically present in inflammatory bowel diseases (IBD) [
6].
While IBD does determine alterations of gut permeability, the so-called “leaky gut”, thus explaining the presence of endotoxemia in this group of persons, it is notable that LPS has been identified as a promoter of different inflammatory pathways also involved in the pathogenesis of IBD. In addition, dysbiosis associated with IBD has been associated with increased production of LPS. It appears that the interaction between LPS and IBD is, in other words, bidirectional rather than monodirectional: while IBD promotes leaky gut and endotoxemia, LPS promotes inflammation, which is pivotal in IBD. The interaction between LPS and promotion and progression of IBD is driven by immunologic pathways and microbiota modulation, which will be discussed below.
2. LPS and IBD, a Complex Crosstalk
LPS is a widely studied inflammatory molecule. Its role in determining severe sepsis is well known and, even though antibiotics have undermined its dangers, it is still worth noting that endotoxemia is associated with a 20% increase in mortality when compared to Gram-positive sepsis [
49]. LPS can also determine a chronic inflammatory status, which has been linked to a wide number of conditions. In these cases, LPS is usually produced by the GM and absorbed in the gut [
4], which appears to be a negative prognostic factor in the case of endotoxemia, given that chronic endotoxemia seems to determine more severe outcomes of acute forms [
50]. Chronic LPS exposure has also been linked to the development of other types of disorders, for instance Alzheimer’s disease [
51].
Once again, systemic immune alterations follow more localized changes, thus LPS also directly promotes gut inflammation. It has been observed, for instance, that LPS promotes extracellular matrix protein (ECM)-1 expression in intestinal macrophages, a situation characteristic of Crohn’s disease and ulcerative colitis [
52]. LPS also induces the expression of inflammatory IL, such as IL-8, and directly damages the epithelial barrier [
53].
We will discuss below the different aspects of the interaction between LPS, immunity, and microbiota, then discuss how these different situations may affect the development of IBD and its progression.
2.1. LPS and the Immune System
LPS interacts mainly with the innate components of the immune system. The key component of this interaction is the TLR4, which triggers the expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB), through the Myeloid Differentiation Primary Response Gene 88 and TIR Domain-Containing Adaptor Protein [
54]. NFkB is crucial to starting a variety of different inflammatory responses, mediated by tumor necrosis factor (TNF)-α and IL-12, which in turn further stimulates the NFkB pathway [
55]. At the same time, the interaction between LPS and TLR4 also triggers the release of IFN-β. All these interactions stimulate the differentiation of inflammatory CD4 subtypes (particularly, Th1 and Th17). Interestingly, while APCs are the most common immune cells which present TLR4 expression, B lymphocytes can also express it if stimulated by specific molecules such as IL-4, which is produced by Th2 cells, another inflammatory subset of CD4+ lymphocytes [
56]. B-lymphocytes stimulated by LPS increase their production of IL-6, TNF-α and increase their antibody production, as seen in the case of
Francisella tularensis [
57].
Overall, TLR4 induces inflammatory responses from the adaptive immune compartment through different molecules including IFN-γ, which is also involved in the activation of signal transducer and activator of transcription 1 (STAT1) [
58]. The activation of this pathway further enhances inflammation. NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome is also activated in monocytes and induces IL-1β and IL-18 expression, critical for promoting the activation of other inflammatory molecules, such as TNF-α [
59].
However, LPS stimulation of T cells can, in some subsets, instead induce an anti-inflammatory reaction: in vivo experiments in mice have shown that, in an asthmatic subset, LPS stimulation might benefit inflammation levels, improving the Th1/Th2 ratio [
60]. While in patients with asthma, the reduction in inflammation can be a benefit, LPS appears to reduce inflammatory patterns in T lymphocytes also in other contexts. For instance, in viral/bacterial coinfection, LPS has been seen to reduce the number of T cells recognizing the virus, particularly natural killers (NK), suggesting that TLR-4 may actually play an important role in viral-bacterial coinfections [
61].
TLR4 is also expressed at an endothelial level, where it can activate apoptosis and trigger the expression of other inflammatory cytokines [
62]. In those affected by IBD, both Crohn’s disease and ulcerative colitis, TLR4 is more expressed than in healthy individuals, thus LPS activity is significantly amplified in this context [
56].
A summary of the interactions between immunity and LPS can be found in .
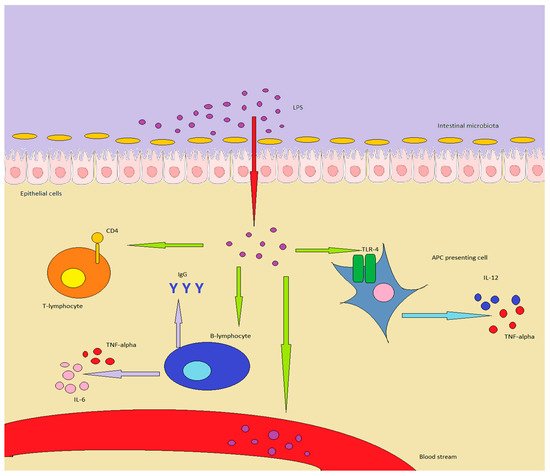
Figure 1. LPS in healthy individuals is produced by GM or enters through food. In inflammatory conditions or in a “leaky gut”, it can overcome the epithelial barrier, determining important effects on the immune system: in particular, APC are stimulated to produce TNF-alpha and IL-12. B-lymphocytes increase antibody production, while also producing TNF-alpha and IL-6, while T-lymphocytes are shifted towards a Th1/Th17 expression pattern. Finally, LPS can enter the bloodstream, inducing chronic or acute endotoxemia.
2.2. LPS and Microbiota
Microbiota is the main source of LPS in healthy individuals. However, high doses of LPS are quite well-tolerated, thus it seems that LPS is relevant only once the inflammation has begun. However, this information could be partial: it has been observed that different bacteria produce different types of LPS, some of which are more likely to determine an inflammatory response. An example of this is the differences between
Bacteroides’ LPS, which is rather harmless, while LPS produced by
E. coli is highly toxic. The consequences of this different type of exposure have been studied in Finnish, Russian, and Estonian children, and
Bacteroides-produced LPS seems to act as an “immune-educator” in the gut [
63]. LPS produced by
E. coli also increases the levels of fecal calprotectin, which is a marker of intestinal inflammation [
64]. TLR4 in cells in the intestine is not activated by LPS of typically commensal bacteria, but the same cannot be said for other organs and systems [
63].
The different composition of the GM is, in other words, one of the driving factors in determining whether LPS is toxic. It is not merely LPS-producing bacteria that determine this, but also other components of the GM. One possible explanation is that certain toxic pathogens could determine a leaky gut, thus allowing even less harmful LPS to enter the bloodstream and trigger non-gut-associated cells [
65].
In addition, LPS can be introduced in the gut through food: milk, for instance, can contain high quantities of it, which can determine a TLR4-driven reaction: in this case, rather than witnessing a microbiota-drive LPS activity, we observe the exact opposite, with LPS capable of determining gut inflammation, which in turn drives changes in GM [
65].
Some authors suppose that orally ingested LPS is not capable of determining such damaging effects, suggesting that through this route of administration LPS is not harmful. Yet, these studies were focusing on pre- and pro-biotics, thus the results are not as widely appliable as one could hope [
66].
One aspect among which most studies agree on is that, whether it be ingested orally or directly produced by one’s GM, LPS toxicity is highly dependent on dietary factors.
Western diet has been associated with the risk of developing metabolic endotoxemia, with data showing that a meal rich in fat can produce a significant increase in LPS blood levels [
67]. On the other hand, a diet rich in fruit and vegetables, on the model of the Mediterranean diet, is instead associated with chronically lower LPS levels.
It is difficult to identify exactly the nature of the interaction: on the one hand, it does seem that the impact on LPS levels may be directly dependent on the diet, which could, in turn, mean that either some foods contain high levels of LPS, or that some foods induce an increased LPS production; on the other hand, an unhealthy diet may simply trigger GM alterations, which increase overall LPS production [
68,
69].
Both explanations are likely true to some extent, thus diet probably contributes both directly and indirectly to LPS induced endotoxemia.
The crosstalk between LPS and microbiota is also interesting in the context of IBD: indeed, the modifications described until now in individuals developing endotoxemia are the same ones identified in those who suffer from ulcerative colitis and Crohn’s disease [
34]. Furthermore, LPS acts as a bridge between the GM and immunity: in particular, LPS can trigger inflammatory activation of intestinal macrophages, which shift their cytokine production from IL-10 to IL-1, IL-6, IL-8, and TNF-α [
70], which impacts the immune system on a larger scale too [
71,
72].
2.3. LPS and IBD, What Do We Know
It appears clear that LPS acts as a potent immunomodulator, with interesting implications as far as the pathogenesis and progression of the disease.
As discussed, LPS acts both on the adaptive and innate immune system. In particular, it has the capacity to activate different immune pathways also involved in the pathogenesis of ulcerative colitis and Crohn’s disease. Macrophage polarization is among them: similar to T-lymphocytes, macrophages can exhibit different properties and cytokine production properties. M1 macrophages exhibit an inflammatory profile, consisting of TNF-α, IL-1α, IL-1β, IL-6, IL-12, CXCL9, and CXCL10 production, and are triggered by LPS and Th1 signaling molecules [
73]. M1 polarization is also regulated by NF-kB, STAT1, STAT5, IRF3, and IRF5 pathways. The presence of M1 macrophages in the gut is typically found in both Crohn’s disease and ulcerative colitis, and LPS stimulation, as seen in murine models, is important in further promoting their expression and local inflammation [
52].
M1 polarization also determines gut permeability alterations: the TLR4/NF-κB and JAK/STAT3 signaling pathways, for instance, are activated in this scenario, promoting epithelial alterations, leading to a leaky gut scenario. The blockade of this pathway does not only improve gut permeability, but also has a positive impact on persons with Crohn’s disease and ulcerative colitis, with reduced inflammation and flares [
74]. In vitro models also demonstrated that interrupting the LPS signaling pathway improves the conditions of the epithelial barrier, through NF-kB inhibition [
53].
While LPS promotes M1 polarization, some studies suggest that it may be important in stimulating autophagy in macrophages, which appears to play an important role in preventing IBD, particularly Crohn’s disease, for instance reducing levels of reactive oxygen species [
75].
Immune system dysregulation is also important in the pathogenesis of both ulcerative colitis and Crohn’s disease, and LPS can stimulate its progression. LPS can indeed determine chronic low-grade systemic inflammation through the mechanisms of endotoxemia. It has been observed, for instance, that in persons chronically exposed to high levels of LPS, it is possible to find not only increased LPS levels but even LPS-positive bacterial extracellular vesicles. Patients with altered gut permeability at particularly high risk [
6].
Also, LPS can promote the expression of fecal calprotectin, a direct consequence of intestinal inflammation. An increase in the levels of fecal calprotectin is also typical of Crohn’s disease and ulcerative colitis [
76,
77].
Another aspect that is worth noting is that systemic LPS induces the expression of IL-6 and other inflammatory cytokines, which can promote Crohn’s disease and ulcerative colitis [
71].
Microbiota also promotes the interaction between IBD pathogenesis and progression and LPS: indeed, different bacteria present in the microbiota can produce LPS, while also directly damaging the epithelial barrier [
70].
As discussed above, LPS is produced by the gut microbiota, but can also come from exogenous sources. While in normal conditions LPS does not cross the gut barrier, it has been observed that it can enter the bloodstream in conditions of altered permeability, such as Crohn’s disease and ulcerative colitis [
78]. Furthermore, the presence of LPS-producing bacteria also helps alter gut permeability, resulting in a vicious circle of self-promoting inflammation [
79].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22126242