Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Anatomy & Morphology
After ingestion of food, the gastrointestinal (GI) tract is activated to facilitate transport, digestion and absorption of nutrients. Regional differences exist within the GI tract with respect to the modulation of these processes. Entry of nutrients into the small bowel activates so-called “intestinal brakes”, negative feedback mechanisms that not only affect motility and secretion but also appetite and energy intake.
- intestinal brake
- satiation
1. Nutrient Sensing in the Gut
Signals mediating satiety and satiation arise from various locations within the luminal gastrointestinal tract including the stomach, duodenum, jejunum, ileum and colon [1]. Ingestion of food results in mechanical stimulation by distension of the stomach and small intestine and in chemical stimulation via activation of nutrient receptors on enteroendocrine cells (EECs). These EECs play a pivotal role in the gastrointestinal and central regulation of not only of gastrointestinal (GI) motility and secretion but also of food intake. ECCs are scattered as single cells throughout the intestinal tract, located within the intestinal crypts and villi, and comprise about 1% of the total epithelial cell population. EECs act as sensors of luminal content, especially of nutrients, and function as trans-epithelial signal transduction conduits with apical physiochemical signals resulting in basolateral release and exocytosis of biological mediators. Nutrients or their breakdown products interact with G-protein coupled receptors (GPCRs) on EECs resulting in the secretion of gastrointestinal peptides such as cholecystokinin (CCK), peptide YY (PYY) and glucagon-like peptide-1 (GLP-1). These mediators either act in a classical endocrine fashion or by a paracrine effect on adjacent cells, including vagal afferent fibers. Non-nutrient chemical factors also regulate EEC activity, for example via sensing of tastants such as bitter, sweet, salt, sour and umami.
EECs carry specific receptors that upon sensing activate intracellular pathways either through direct gating of ion channels such as the sodium-dependent glucose co-transporter 1 (SGLT-1) or via activation of GPCRs. In recent years, various GPCRs have been identified such as sweet taste receptors (TAS1R/TAS2R), fatty acid-sensing receptors (GPR40, GPR43, GPR119 and GPR120) and various other types including PPAR, melanocortin, TRP family and opioids [2][3][4][5].
G-protein coupled taste receptors are expressed not only on the human tongue but also in stomach, proximal and distal small intestine and colon. Bitter taste is sensed by the TAS2R receptor while the TAS1R receptor family is triggered by sweet and umami. Taste receptors are able to “taste” luminal content and transmit signals that induce the release of GI peptides, thereby influencing satiety and food intake in humans [6].
2. Gastric Satiation Signals
Apart from its function to store food, to mix and grind stomach content and initiate the process of digestion, the stomach is able to monitor “food ingestion”. Consensus exists on the important role of gastric mechano-sensation in the regulation of satiety and food intake. In contrast to intestinal satiation, which is merely nutrient-induced, gastric satiation is merely volume-dependent. This has been shown for the first time in pyloric cuff experiments in rats [7]. In these experiments, saline or nutrient solutions were infused into the stomach but the pyloric cuffs prevented the infusate from entering the duodenum. Both the saline and nutrient infusate resulted in a similar reduction in food intake, showing that the nutritive effect did not add to the volumetric effect [7]. The satiating effect of gastric distension has been confirmed in human studies employing intragastric balloons. Prolonged distension of gastric balloons is known to result in reduction of food intake and subsequent weight loss [8].
3. Intestinal Satiation Signals
Exposure of the small intestinal lumen to nutrients induces satiety and a reduction in food intake. This was first observed in animal studies using gastric fistulas to exclude ingested food from entering the small intestine [9]. The animals would eat continuously when food was drained from the stomach. When the gastric fistula was closed and food entered the small intestine, the animals rapidly stopped eating, pointing to the pivotal role of the intestine in inducing satiety and satiation. Later, in vivo human intubation studies revealed that intestinal perfusion of proteins, carbohydrates or lipids, all resulted in a significant increase in satiety and decrease in food intake [10][11][12][13].
The GI peptides CCK, GLP-1 and PYY, secreted from EECs are known as mediators of intestinal satiation. These peptides induce their effects either via entering the bloodstream, acting as hormones (endocrine effect), via activation of vagal afferents (neuronal effect) or via an effect on neighboring cells (paracrine effect). Ghrelin is produced from the stomach and currently is the only GI peptide known to increase food intake by accelerating gastric emptying [1][6][13][14][15].
4. Intestinal Brakes
The process of motility, secretion, digestion and absorption is activated upon ingestion of food and its transport into stomach and duodenum. Thereafter, the appearance of nutrients further downward in the small intestine, during the process of digestion, results in activation of the so-called ”intestinal brakes”: feedback mechanisms from different parts of the intestine to the stomach, to the more proximal parts of the small intestine and also to the central nervous system.
Entry of nutrients into duodenum or jejunum activates the “duodenal brake” or “jejunal brake” while infusion of nutrients into the ileum activates the more distal “ileal brake”.
The “duodenal brake” has been well documented as feedback from the duodenum to regulate gastric physiology, that is gastric acid secretion and gastric emptying. Inhibition of gastric emptying is a crucial “brake” against delivery of nutrients to the intestine in excess of digestive and absorptive capacity. In humans, gastric emptying is slowed in proportion to the energy density of the meal, thus leveling the rate of energy delivery to the duodenum [1][7][8][11].
The “ileal brake” is a negative feedback mechanism from the more distal to the proximal gastrointestinal (GI) tract that brings the process of transport, digestion and absorption of nutrients to an end (Figure 1). Activation of the “ileal brake” results in a reduction of gastric acid, biliary and pancreatic secretion, with inhibition of gastric emptying, intestinal motility and transport [14]. This concept of more distal intestinal brake activation with proximal inhibition of motility and secretion was derived from ileal transposition studies in rats. Koopmans and Sclafani were the first to show that transposing a segment of ileum to more proximal regions of the small intestine (i.e., duodenum) also resulted in a significant reduction in food intake and was associated with weight reduction in rats [15]. It was hypothesized that the hormonal changes induced by ileal transposition may have resulted in the observed reduction in food intake and increase in weight loss. Indeed, Strader et al. [16] observed that ileal transposition resulted in 3–4 times higher serum GLP-1 and PYY levels with an increase in satiety and weight loss that was proportional to the measured serum levels of GLP-1 and PYY.
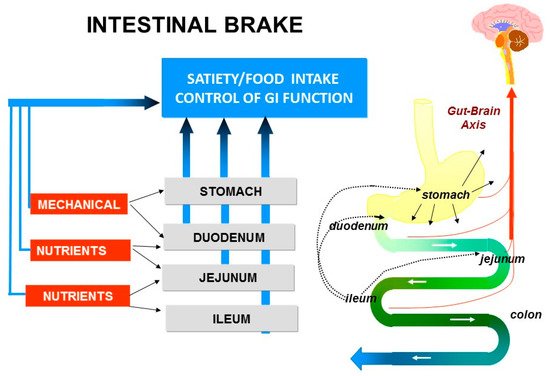
Figure 1. Intestinal brake: effect of luminal stimulation in stomach, duodenum, jejunum and ileum on neurohormonal control of gastrointestinal function, food intake and satiety.
Such a feedback inhibitory mechanism from the distal to the proximal GI tract has repeatedly been demonstrated in animal models. In the 1980s, the first human studies showed that infusion of fat or protein in the ileum delayed gastric emptying and intestinal transport and also increased feelings of satiety and reduced food intake. Welch et al., Read et al. and Spiller et al. [10][11][12][13] were among the first to point to the potent anorexic effect of “ileal brake” activation in humans through intestinal nutrient infusion. Welch et al. showed that an ileal lipid infusion of 370 kcal resulted in a decrease in food intake of 575 kcal, resulting in a net reduction in intake of 205 kcal. In a subsequent study comparing jejunal fat versus ileal fat infusion, the anorexic effect was more pronounced when fat was infused into the jejunum instead of the ileum [10]. One should consider that in these studies very high amounts of fat of up to 41 g were administered intestinally. These supraphysiologic amounts may have caused spilling of fat to more distal intestinal regions resulting in larger areas with activated nutrient receptors.
This entry is adapted from the peer-reviewed paper 10.3390/nu13051601
References
- Cummings, D.E.; Overduin, J. Gastrointestinal regulation of food intake. J. Clin. Investig. 2007, 117, 13–23.
- Burdyga, G.; Lal, S.; Varro, A.; Dimaline, R.; Thompson, D.G.; Dockray, G.J. Expression of Cannabinoid CB1 Receptors by Vagal Afferent Neurons Is Inhibited by Cholecystokinin. J. Neurosci. 2004, 24, 2708–2715.
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94.
- Lauffer, L.M.; Iakoubov, R.; Brubaker, P.L. GPR119 Is Essential for Oleoylethanolamide-Induced Glucagon-Like Peptide-1 Secretion from the Intestinal Enteroendocrine L-Cell. Diabetes 2009, 58, 1058–1066.
- Tanaka, T.; Katsuma, S.; Adachi, T.; Koshimizu, T.-A.; Hirasawa, A.; Tsujimoto, G. Free fatty acids induce cholecystokinin secretion through GPR120. Naunyn Schmiedeberg’s Arch. Pharmacol. 2007, 377, 523–527.
- Depoortere, I. Taste receptors of the gut: Emerging roles in health and disease. Gut 2014, 63, 179–190.
- Phillips, R.J.; Powley, T.L. Gastric volume rather than nutrient content inhibits food intake. Am. J. Physiol. 1996, 271, R766–R769.
- Powley, T.L.; Phillips, R.J. Gastric satiation is volumetric, intestinal satiation is nutritive. Physiol. Behav. 2004, 82, 69–74.
- Gibbs, J.; Young, R.C.; Smith, G.P. Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature 1973, 245, 323–325.
- Welch, I.M.; Sepple, C.P.; Read, N.W. Comparisons of the effects on satiety and eating behaviour of infusion of lipid into the different regions of the small intestine. Gut 1988, 29, 306–311.
- Read, N.W.; McFarlane, A.; Kinsman, R.I.; Bates, T.E.; Blackhall, N.W.; Farrar, G.B.; Hall, J.C.; Moss, G.; Morris, A.P.; O’Neill, B.; et al. Effect of infusion of nutrient solutions into the ileum on gastrointestinal transit and plasma levels of neurotensin and enteroglucagon. Gastroenterology 1984, 86, 274–280.
- Spiller, R.C.; Trotman, I.F.; Higgins, B.E.; Ghatei, M.A.; Grimble, G.K.; Lee, Y.C.; Bloom, S.R.; Misiewicz, J.J.; Silk, D.B. The ileal brake inhibition of jejunal motility after ileal fat perfusion in man. Gut 1984, 25, 365–374.
- Welch, I.; Saunders, K.; Read, N. Effect of ileal and intravenous infusions of fat emulsions on feeding and satiety in human volunteers. Gastroenterology 1985, 89, 1293–1297.
- Layer, P.; Peschel, S.; Schlesinger, T.; Goebell, H. Human pancreatic secretion and intestinal motility: Effects of ileal nutrient perfusion. Am. J. Physiol. Liver Physiol. 1990, 258, G196–G201.
- Koopmans, H.S.; Sclafani, A. Control of body weight by lower gut signals. Int. J. Obes. 1981, 5, 491–495.
- Strader, A.D.; Vahl, T.P.; Jandacek, R.J.; Woods, S.C.; D’Alessio, D.A.; Seeley, R.J. Weight loss through ileal transposition is accompanied by increased ileal hormone secretion and synthesis in rats. Am. J. Physiol. Metab. 2005, 288, E447–E453.
This entry is offline, you can click here to edit this entry!