Human midbrain dopamine (DA) neurons are a heterogeneous group of cells that share a common neurotransmitter phenotype and are in close anatomical proximity but display different functions, sensitivity to degeneration, and axonal innervation targets. The A9 DA neuron subtype controls motor function and is primarily degenerated in Parkinson’s disease (PD), whereas A10 neurons are largely unaffected by the condition, and their dysfunction is associated with neuropsychiatric disorders. Currently, DA neurons can only be reliably classified on the basis of topographical features, including anatomical location in the midbrain and projection targets in the forebrain.
1. Introduction
Dopamine (DA) neurons in the ventral midbrain (VM) constitute a heterogeneous group of cells with different anatomical locations, physiological properties, and projection patterns. These cells are involved in a broad spectrum of cerebral functions associated with voluntary movement, as well as with cognitive and emotive tasks [
1,
2].
DA neurons are traditionally divided into three subtypes. A9 neurons are located in the substantia nigra pars compacta (SNpc), A10 in the ventral tegmental (VTA) area, and A8 in the retrorubral field. As well as displaying different functions and axonal innervations, they exhibit heterogeneous susceptibility to disease processes and show fundamental differences in vulnerability to cell death in Parkinson’s disease (PD). Each cell group forms specific connections and projects to distinct anatomical target areas of the central nervous system (CNS), establishing separately controlled functional networks [
3,
4,
5]. A8 and A10 neurons innervate the ventral striatum, nucleus accumbens, septum, and the prefrontal cortex via the mesolimbic pathway, and are mainly involved in controlling emotional behavior and motivation. A9 neurons project to the striatum, forming the nigrostriatal pathway that regulates motor function. They also have different functional and projection patterns, as compared to the other subtypes. A9 neurons in humans and other primates also show accumulation of the neuromelanin pigment. A9 neurons are primarily degenerated in PD, making them the subject of more extensive studies [
6,
7,
8,
9,
10].
Given the key role of A9 cells in PD, the generation of this subtype of DA neurons from stem cell sources is an area of intense investigation ultimately aimed at exploiting their use in cell-based replacement treatment. For decades, neuroscientists have attempted to identify selective markers expressed in these subpopulations, in order to dissect the complexity of DA regulatory networks and design more effective therapeutic strategies [
11,
12]. The inaccessibility of fetal and adult human brain tissue makes it difficult to elucidate the relation between histological assessments and the heterogeneity of DA neurons at the molecular level. The compilation of a comprehensive dataset linking the molecular diversity of DA neurons with their function and anatomical innervation target would require a systematic genome-wide molecular classification at single-cell resolution [
13,
14].
The advent of single cell sequencing technologies has provided unprecedented insights into DA subtypes and uncovered an unexpectedly high heterogeneity, even within anatomically defined DA subgroups. Such approaches have already been used in the adult mouse brain to unbiasedly catalog DA neurons, based on their gene expression profiles. However, the question of whether a similar diversity exists in the human midbrain and whether molecularly distinct DA subtypes correspond to innervation target regions and the traditional classification based on anatomical landmarks, remains completely unexplored [
3,
15,
16].
The ability to recreate human neurons from human pluripotent stem cells (hPSCs) opens up exciting opportunities to study human neurogenesis and understand mechanisms and treatments for brain disease(s). It also provides access to a renewable source of cells potentially suitable for therapeutic applications, including drug screening and cell-based therapy () [
17,
18]. The generation of human tissues in vitro, combined with the use of human cells in xenograft transplantation models and sophisticated transcriptomics technologies, is ushering in the new era of “human” biology. Breaking down the intricate regulatory system controlling DA neuron subtype specification into its individual layers will provide crucial new insights into the transcription factors and molecular cues that specifically drive the diversity of human DA neurons [
19,
20,
21].
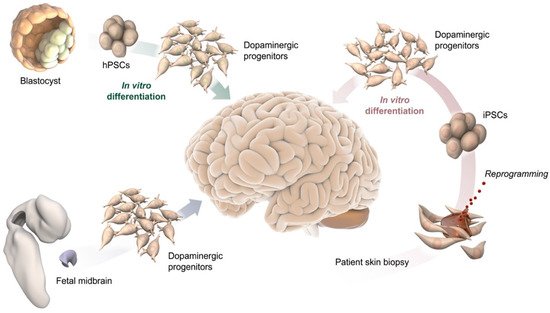
Figure 1. Schematic illustration of potential DA progenitor sources for modeling VM differentiation and for therapeutic application in PD, including blastocysts and their pluripotent stem cell derivative (top-left), fetal midbrain tissue (bottom left), and somatic cells from adult individuals, such as a skin biopsy used to reprogram cells to pluripotency (bottom-right).
2. Single Cell Sequencing in Decoding Human Brain Complexity
Major advances in next-generation sequencing technologies mean that it is now possible to characterize the genome, epigenome, and transcriptome at single-cell resolution [
13,
90]. Single-cell RNA sequencing (scRNAseq) has emerged as a powerful methodology that is able to profile the full transcriptome of individual cells, providing an unprecedented measurement of heterogeneity, an intrinsic feature of all living biological systems [
91]. scRNAseq is able to deconstruct the complexity of fundamental biological processes, by characterizing heterogeneous and rare cell populations and identifying novel cell types and regulatory gene networks, thereby elucidating the molecular mechanisms controlling human brain functions. For the analysis of brain tissues, cell dissociation is the most critical step, as conditions in this phase directly affect the molecular profiles of cells. In neuroscience, single nuclei RNAseq is rapidly replacing scRNAseq, since neurons are hard to capture using single-cell dissociation protocols [
92,
93]. Several technologies for library preparation based on microdroplet technology are reported, such as 10X genomics and Drop-Seq [
94]. In these methods, a cell/nucleus, reaction enzymes, and a barcoded bead are encapsulated in an oil droplet. Reverse transcription, including tagging each RNA with a cell-specific barcode takes place within each droplet. As an alternative to droplet-based technologies, microwell protocols, including Smart-Seq, can be deployed. Although these protocols generally have lower throughput, they are able to capture full-length transcripts and have a better sensitivity for detecting lowly expressed genes [
95,
96]. More recently, combinatorial single cell indexing platforms, based on split-and-pool DNA-barcoding strategies, offer another highly scalable approach comparable to the droplet systems, but without the need for specific instruments. The large number of cells that can now be sequenced combined with the relative sparsity of data, has posed unique data science challenges, including handling of data sparsity, robust differential expression, and accurate trajectory inference.
For years, scientists have been intrigued by the fact that DA neurons, which make up less than 1% of the total number of brain neurons, can play such diverse roles in influencing a large spectrum of cognitive and motor functions. With the advent of single-cell technologies, research efforts have aimed to create a systematic catalog of molecularly distinct DA neuron subtypes exhibiting specific physiological and functional properties, in order to understand their different vulnerability in PD and to develop more effective pharmacological and cell-based treatments [
15,
49,
91].
Recent studies examined DA neuron subtypes in mouse and humans, based on their molecular profiles at single-cell resolution. In transgenic adult mice, isolation based on Pitx3 expression and the subsequent sequencing of single neurons, identified five molecularly distinct DA clusters in the midbrain. It exhibited the different gene expression levels of dopamine-producing machinery components, suggesting that each one has a distinct role in mammalian VM [
97]. Similarly, a single-cell dataset comparison between fetal VM dissected from human embryos (6–11 weeks) and mouse, captured the similarities and differences in VM development between the two species [
26]. The findings of this investigation revealed the evolutionary conservation of cell type diversity in humans and mice, highlighting the major differences in proliferation and timing of DA development. However, despite recent efforts, classifying neurons into subtypes remains far from straightforward. All the subgroups are closely related, as they all display a typical DA neuron signature expressing the genes required for DA synthesis but possess unique molecular features with a small set of differently expressed genes. To identify rare subtypes, a large number of cells need to be captured in order to ensure sufficient representation, especially considering that DA neurons make up only a tiny fraction of the cells captured when sequencing the mammalian midbrain. At the same time, techniques with high throughput (e.g., 10×, sci-fi-seq) have lower sensitivity than, for example, Smart-seq, meaning that a delicate decision has to be taken on whether to focus on the number of analyzed cells versus sensitivity. Another complicating factor is that the transcriptome alone might not be sufficient to define closely related DA subtypes. This limit can be overcome by using multi-omics approaches, discussed in detail in the following subsection.
3. Advances in Single Cell Transcriptomics
scRNAseq is now being combined with morphological characterization, lineage tracing, and functional analysis, in order to compile a comprehensive catalog of molecularly distinct neuronal subtypes, a valuable asset for elucidating their development and function in human brain [
98].
4.1. Spatial Transcriptomics
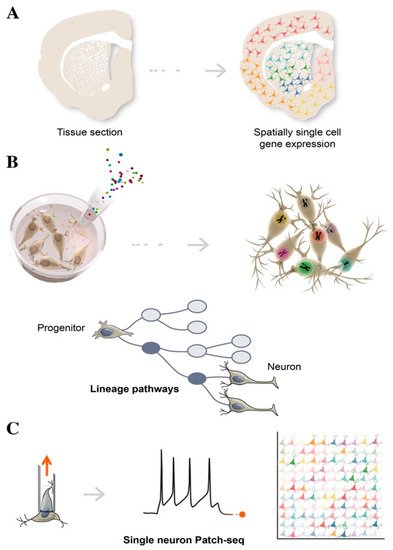
Spatial transcriptomics is used to better understand the function of a single cell in distinct spatial architectures within a brain tissue sample, as well as to determine the subcellular localization of RNAs (A) [
99,
100]. Spatial transcriptomics is a revolutionary technology and is based on the assumption that in order to define the function of specific cell types, it is necessary to obtain positional information while preserving cell–cell communication and intracellular regulation. A tissue section is placed on an array containing probes, to capture gene expression data. This results in a topologically-based transcriptional deconvolution of human tissue, which is required to provide a molecular identity map illustrating how cell types are localized within the tissue. Although spatial transcriptomics does not require cell dissociation, thus, providing a better understanding of the role of an individual cell within its microenvironment, sequencing resolution is significantly lower than in conventional single-cell approaches and is also highly dependent on the structure of tissues and the harvesting procedures [
101].
Figure 3. Schematic illustration of single-cell technologies combining transcriptomics with morphological features, to assign cell types to their locations in histological sections (A), with developmental lineage tracing to track cellular relationships during development or regeneration (B), and with functional analysis to simultaneously profile single-cell transcriptome and electrophysiological properties (C).
3.2. Cellular Barcoding
Lineage tracing tracks the diversification history of cellular subtypes generated during development. This technology labels an individual cell by introducing heritable genetic makers called “barcodes” at early time-points, in order to trace the state of its clonal progeny at different stages of stem cell differentiation, via sequencing (B) [
102]. Barcoding is a lentiviral cell tag method usually consisting of a 5–7 bp fixed region and a 10–12 bp variable region, which represent a unique identifier for individual cells. A combination of 4–5 tags can be used to increase barcoding library tracking. By harvesting cells at different stages, single cell computational inference analysis is able to place individual cells along a temporal axis, reconstructing a developmental continuum of stem cell states from cell origin to differentiated subtypes, including intermediate progenitors [
103]. To reconstruct a composite phylogenetic tree that accurately defines branch points and sister cells from each division, single-cell chromatin accessibility assays such single-cell ATAC-seq can be adopted to add genomic and epigenomic details of these heritable properties. Reconstructing lineage history and mapping progeny relationships deduces cell fate decisions toward specific neuronal subtypes, and provides valuable insights into how to best direct and refine stem cell differentiation for use in biomedical applications [
104]. By employing machine learning and gene regulatory network analysis, it is becoming increasingly possible to identify new regulators of differentiation that trigger a cascade of transcriptional events, leading to cell populations with a functionally mature phenotype.
3.3. Multimodal Single Cell Data
Multimodal single-cell data transcriptomics alone is often unable to separate molecularly similar, but functionally distinct, neuronal cells. Multimodal single-cell technologies, which simultaneously profile multiple data types in the same cell represent the latest frontier in the discovery and characterization of cell states [
105,
106]. For example, utilizing oligonucleotide-tagged antibodies, in conjunction with scRNAseq, a method called CITE-seq, was used to simultaneously quantify RNA and surface protein abundance in single cells, via the sequencing of antibody-derived tags [
107,
108]. Recent advancements now enable the simultaneous profiling of transcriptome, alongside either chromatin accessibility, methylation, or nucleosome occupancy. Combined analysis of single-cell transcriptomes and proteomics has proven more challenging, as these are vastly different molecular modalities. Converting them into a uniform state that can be analyzed together in an “omics-wide” manner is a substantial technical challenge.
3.4. Patch-Seq
Patch-seq is a specific example of multimodal single-cell sequencing, combining electrophysiological whole-cell patch-clamp recordings, scRNAseq, and morphological characterization, in order to obtain the functional classification of neuronal subtypes (C) [
109]. Of note, single-cell Patch-seq is able to correlate the molecular features of diverse neuronal types with their physiological and functional role in the brain. After electrophysiological recordings, the cell contents are aspirated through the patch-clamp pipette and are prepared for scRNAseq. This technology represents an important step forward in providing high-resolution analysis of rare cell types or a specific subset of neurons, which would otherwise be lost among the thousands of single cells, when using conventional scRNAseq approaches [
110,
111]. Single-cell analysis of electrically dysfunctional neurons is also extremely important for studying the etiology of neurological disorders. Healthy and diseased iPSCs have been widely used to model brain diseases, in order to explore aberrant phenotypes of specific neuronal populations, using single cell Patch-seq.
This entry is adapted from the peer-reviewed paper 10.3390/cells10061366