Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Pre-mRNA splicing is an essential step in gene expression and is catalyzed by two machineries in eukaryotes: the major (U2 type) and minor (U12 type) spliceosomes. While the majority of introns in humans are U2 type, less than 0.4% are U12 type, also known as minor introns (mi-INTs), and require a specialized spliceosome composed of U11, U12, U4atac, U5, and U6atac snRNPs. The high evolutionary conservation and apparent splicing inefficiency of U12 introns have set them apart from their major counterparts and led to speculations on the purpose for their existence.
- minor introns
- U2 introns
- U12 introns
- minor spliceosome
- RNA splicing
1. RNA and Splicing
The view that RNA is simply an intermediate between DNA (the keeper of genetic information) and proteins (the executioners of all cellular functions) has long been challenged by the discoveries that gave RNA its own catalytic functions such as self-splicing introns [1] and the ribonuclease P catalyst [2]. These discoveries provided further evidence for the RNA world theory, which stipulates that life on earth started with RNA. The primary living substance on primitive earth is therefore thought to be RNA or a chemically similar structure, a process that may have started 4 billion years ago. The evidence for this 50 year old theory has grown over the years, and the importance of RNA has grown with it, giving rise to new fields in RNA biology that have expanded toward medical/clinical applications, including experiments done in the early 1990s showing protein production from an mRNA injected into mice skeletal muscle [3] and antibody generation from influenza virus mRNA injection [4], to most recently using mRNA as a vaccine against SARS-CoV-2 [5,6]. Thus, RNA-based therapies are quickly progressing to treat many ailments by targeting various pathways in RNA metabolism, including pre-mRNA splicing, which has garnered increasing attention after a handful of successful applications.
Pre-mRNA splicing was initially discovered in adenoviruses but was later described to occur in all eukaryotes [7]. More than 95% of genes in humans are transcribed as pre-mRNAs containing introns (intervening regions) that interfere with the required sequence continuity of exons (expressed regions) [7,8]. Splicing occurs co-transcriptionally to ensure the removal of introns and subsequent joining of exons, making it an essential and required step for the proper formation of a mature mRNA transcript that could be used as a template for protein synthesis [7,8]. Of note here is that a large number of intron-containing genes are transcribed and spliced yet do not get translated into a protein but rather have key functions as noncoding RNAs. Broadly, the sequences involved in splicing include a 5′ splice site (5′ ss), a branch point sequence (BPS), and a 3′ splice site (3′ ss), and splicing is carried out as a two-step reaction, starting with 5′ splice site cleavage, generating a lariat, and ending with 3′ splice site cleavage and exon ligation [8]. This reaction is catalyzed by the spliceosome, an RNA-protein complex that requires ATP and Uridyl-rich small ribonucleoproteins (U snRNPs) [9].
2. The Other Side of the Splicing Coin—An Overview
The majority of intron splicing relies on the ubiquitous major spliceosome formed by the U1, U2, U4, U5, and U6 snRNPs [10,11]. These introns are loosely termed U2 introns and are referred to as such hereafter. Interestingly, eukaryotes carry in their genomes a less common type of introns termed minor introns (mi-INTs) or U12 introns (in this review, we use both terms interchangeably). The number of mi-INTs vary between species with humans having one of the largest number (~750) of minor-intron-containing genes (miGs) [12,13,14,15]. The mere existence of mi-INTs is remarkable for many reasons. Rather than using the highly abundant and ubiquitous major spliceosome, mi-INTs utilize a specialized spliceosome composed of a distinct set of snRNPs (U11, U12, U4atac, U5, and U6atac) [12,13,14,15]. The first mi-INTs were identified due to the unusual dinucleotide termini (AU at the 5′ end and AC at the 3′ end of the intron, compared to the conventional GU and AG, respectively) [12,16]. With the discovery of more mi-INTs, it became clear that the AU-AC dinucleotide at the intron boundaries is not what distinguishes U12 from U2 introns as most mi-INTs were shown to have a GU-AG dinucleotide but possess other unique features that set them apart from U2 introns [17,18]. These features played an important role in computationally identifying mi-INTs and include sequences within the introns as well as the conservation of their 5′ ss and BPS over 100 million years [19]. The peculiarity of mi-INTs however extends beyond their sequence features. In spite of their low abundance, the position of mi-INTs in their host genes is also highly conserved between humans, animals, and plants [14,20,21]. On the other hand, miGs tend to show commonality in their functions and have been coined by Burge et al. as “information processing genes” [19]. These functions include DNA transcription, replication and repair, RNA processing and translation, cytoskeletal organization, vesicular transport, voltage gated ion channel activity, and Ras-Raf signaling [22,23]. These very important functions seemed for a long time to be contradictory to in vitro and in vivo experiments showing slow splicing rates and hence inefficiency of the splicing of mi-INTs, making them bottlenecks for the expression of their host genes [24]. Moreover, data showing that mi-INTs can be experimentally converted to U2 introns with relative ease, as this required only a few mutations in their 5′ ss [17,19,25], adding serious doubt regarding the purpose of minor intron splicing altogether. Lastly, another puzzling feature of mi-INTs is the low abundance of their minor spliceosome (~100-fold compared to the major spliceosome), especially the catalytic component U6atac snRNP [26]. The dilemma of these seemingly contradictory features has been partially resolved as discussed later in this review.
Pinpointing the exact origin of introns in general has been elusive. However, their approximate age has been speculated based on their absence from prokaryotic genomes and presence in most eukaryotic and protist genomes [19,27,28]. Prior to the discovery of the U12 splicing pathway, attempts to explain the origin of introns followed two theories: the “exon theory of genes” and the “protosplice theory”. The former stipulated that introns came into existence to allow exon assortment, giving rise to diverse genes through intron recombination, while the latter suggested that introns invaded eukaryotic genomes and were inserted at protosplice sites [29,30,31,32]. The discovery of U12 introns has challenged both theories as the presence of two distinct types of introns had not been accounted for by either theory. mi-INTs have a nonrandom distribution (clustered in information processing genes), and they occur less frequently in the genome. However, despite our lack of understanding of the origin of U12 introns, it was clear that these introns have appeared early in eukaryotic evolution and have been conserved in many organisms, spanning several kingdoms [20,27,33]. These include plants (Arabidopsis thaliana), vertebrates (fish, amphibians, birds, and mammals), insects (Drosophila melanogaster and silkworm Bombyx mori), cnidarians (jellyfish), protists (Acanthamoeba castellanii), and fungi (Rhizopus oryzae). Nevertheless, it appears that mi-INTs have also been lost throughout evolution as they are absent in some fungi (Saccharomyces cerevisiae and Schizosaccharomyces pombe), some protists, and nematode (Caenorhabditis elegans) [19,27]. Some evidence suggests that some U12 introns loss has occurred through conversion to U2 introns [17,19]. Again, in light of U12 introns’ apparent disadvantages (slow kinetics, low abundance of minor spliceosome, etc.) and their ability to potentially easily convert to U2 introns, coupled with the extensive evidence of their conservation, and the essential functions of miGs, the conundrum of why mi-INTs even exist ensued. The working model for the why mi-INTs originated and are extremely conserved is that they play an important biological function within the genes that host them, driving their maintenance within miGs and conservation across organisms.
Several lines of evidence support this model of inefficiency for regulatory purposes and challenged the idea that mi-INTs retention is due to an inherent inefficiency in this splicing pathway. It was shown that the splicing inefficiency of mi-INTs resulting in partial or complete intron retention leads by design to specific outcomes, including degradation of their host transcripts by non-sense-mediated decay (NMD) or the exosome, alternative splicing of the transcript, or translation into a truncated protein. These outcomes are in support of mi-INTs retention being a post-transcriptional mechanism regulating miGs expression. Indeed, transcripts retaining mi-INTs were shown to constitute a pool of pre-mRNAs that is on standby to be expressed when required based on cellular needs. This is determined by the level of U6atac snRNP which is regulated through a MAPK signaling pathway. The effectiveness of this regulatory role of mi-INTs lies in the ability of a single intron to regulate the expression of a whole pre-mRNA. Therefore, the overarching function of mi-INTs in hundreds of miGs is suggested to be an expression “molecular switch” [34]. While the molecular switches model implies an all or none regulation, other data suggested a role for mi-INTs in alternative splicing as shown for the splicing factor Srsf10 [35], which is a member of the SR protein family that regulates alternative splicing. Srsf10 autoregulates its splicing through two competing minor and major 5′ splice sites, resulting in a protein-coding or a noncoding transcript, respectively. This autoregulation leads to a dynamic mechanism where the minor spliceosome activity determines Srsf10 gene expression, and this in turns affects the levels of many other SR proteins. This work highlights a role for the minor spliceosome in regulating alternative splicing of many U2 introns indirectly through regulating the levels of SR proteins in cells [35].
The clinical relevance of splicing is well established, as 90% of disease-causing SNPs lie outside the protein-coding regions and many could have underlying mechanisms that would lead to aberrant splicing [36,37]. Indeed, some models estimate that 62% of disease mutations act by affecting splicing [38]. While it is still unclear what exact proportion of these mutations affects mi-INTs splicing as the targets of these mutations are being uncovered, several known mutations do include components of the minor splicing machinery (snRNPs), mi-INTs splice sites, and loci important for biogenesis and assembly of the minor spliceosome. Such aberrations lead to diseases ranging from neurodevelopmental and neurodegenerative to autoimmune disorders and cancer [39]. While the latter is well established in relation to mis-splicing of U2 introns and less so to that of mi-INTs [37,39], the similarity between the two splicing pathways, in addition to some evidence about alterations of mi-INTs splicing in cancer [40,41,42], provides further support that both U2 and U12 intron splicing may be affected in a similar fashion in cancer.
Mutations within components of the minor intron splicing machinery have been linked to the majority of diseases caused by minor intron splicing alterations. These mutations are predicted to affect hundreds of target genes containing mi-INTs, with the possibility of causing systemic defects in many body tissues. However, despite the ubiquitous nature of these mutations, most of the resulting diseases are tissue specific, indicating the possibility that only a subset of target genes has the potential of causing disease. Fortunately, recent advances in genomics coupled with efforts to understand mi-INTs biology have shed some light on the mechanisms underlying disease tissue specificity and ways to identify target genes. In this review, we discuss this tissue specificity based on our current understanding of mi-INTs splicing and suggest experimental approaches using animal models coupled with CRISPR screens to identify target genes. We also bring to light new advances in the field of mi-INTs through discussing mi-INTs characteristic, the current debate of inefficiency vs. regulation, the fate of retained mi-INTs, and the crosstalk between the minor and major spliceosomes.
3. mi-INTs Sequence Characteristics
Initially described as introns with AU-AC termini (Figure 1A), mi-INTs containing these termini were later found to be only a subtype of all U12 introns [12], and the majority of mi-INTs have GU-AG termini [17,18]. Furthermore, mi-INTs could also have other, albeit much rarer, termini including AU-AG, GU-AU, CU-AC, GG-AG, and GA-AG [17,43]. Consequently, mi-INTs classification had to rely on more specific characteristics to distinguish them from U2 introns that have the canonical GU-AG termini. mi-INTs key sequence features lie in the conservation of nucleotide segments in their 5′ ss and BPS immediately upstream of their 3′ ss [12]. The 5′ ss conserved segment includes 8–9 nucleotides (nts) (A/G)UAUCCUUU, and the BPS includes the conserved sequence UCCUUAAC with fewer variations compared to U2 introns [12,19,44]. The splicing of mi-INTs depends on a well-situated 3′ ss creating a functional constraint on the distance between the BPS and the 3′ ss [44]. Thus, this distance is relatively shorter (10–20 nts) in mi-INTs, with the optimal distance being 11–13 nts [44]. Furthermore, mi-INTs lack a polypyrimidine tract which is commonly found in U2 introns (Figure 1A). The splice sites sequences and their conservation in multiple species can be found at https://genome.crg.es/cgi-bin/u12db/u12db.cgi (accessed on 19 May 2021).
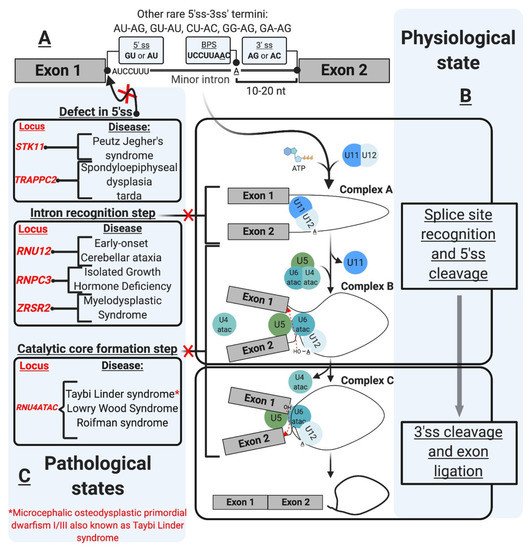
Figure 1. The minor intron splicing pathway. (A). U12-type intron sequence characteristics [1,2,3,4,5,6]. These sequences were initially described in the literature based on DNA nomenclature. These were changed in the figure as the transcript depicted is an RNA transcript. The commonly observed 3′ and 5′ sequences of minor introns are shown in the boxes. The conserved sequences are shown at the 5′ ss and branchpoint (B). Spliceosomal assembly and the splicing reaction [7,8,9,10,11,12] occur as a two-step reaction. The interaction of the preformed U11/U12 di-snRNP leads to the formation of Complex A. U4atac/U6atac.U5 tri-snRNP allows the formation of Complex B, which carries the first transesterification reaction after the release of U4atac and U11. This reaction leads to the formation of Complex C, which carries the second transesterification reaction producing ligates exons and a minor intron lariat. (C). U12-type intron-associated diseases classification [13,14,15,16,17,18,19,20,21]. The diseases are depicted in association with the affected step. Diseases not shown in the figure include amyotrophic lateral sclerosis associated with mutations in fused in sarcoma (FUS) RNA-binding protein [22], and Noonan syndrome due to U12-type intron retention in LZTR1, a regulator of RAS-related GTPases [23].
mi-INTs sequence features are critical for their recognition, the formation/assembly of the splicing machinery, and adequate splicing through the 5′ and 3′ ss (Figure 1B). This splicing is carried out by the less abundant minor spliceosome, with unique snRNPs and protein composition, and slower splicing kinetics (refer to the spliceosome section for more detail).
4. Origin and Evolution: The U12 Splicing Pathway Is as Old as the U2 Pathway
mi-INTs were initially exclusively described in animals [12,16] but were later identified in plants with identical sequence characteristics, dating the origin of the minor spliceosomal pathway to at least one billion years [45], as it is thought to have appeared prior to the divergence of the animal and plant kingdoms. This is supported by the discovery of mi-INTs and the homologous component of the minor splicing pathway (U11, U12 snRNA, and minor spliceosome-specific proteins) in protists and fungi [27]. The presence of components of a minor spliceosomal pathway in protists is evidence that the minor spliceosome is possibly as old as its counterpart, the major spliceosome. The evolution of these splicing pathways could have followed three models: the group II introns parasitic invasion model [19,46,47], the codivergence model [48], or the fission/fusion model [19]. The parasitic invasion model assumes that following endosymbiont invasion of an archaeal organism, descendants of two distinct group II introns invaded nuclear genes sequentially or simultaneously [19,28,46,47]. This may have occurred through repeated lysis of the invading organism (the developing mitochondria) causing seeding of these introns [28,47,49]. The fragmentation of these introns would have led to novel U snRNAs that could utilize proteins of the pre-existing spliceosome (supposedly U2-dependent spliceosome from a prior invasion) to splice these introns [19,47,50,51]. A protosplice site comparison of ancient introns to U2 and U12 type introns in humans and Arabidopsis thaliana suggests that primordial spliceosomal introns were of the U2 type [51]. Consequently, based on the parasitic invasion model, it is thought that U2 introns were the first to populate genes, followed by U12 introns [47,51].
However, this model does not explain the existence of “twintrons” in some pre-mRNAs. A twintron is basically a major intron within a minor intron, such as that in the Drosophila prospero gene, which encodes a protein that plays an important role in axonal outgrowth and cell specification during the development of the nervous system [52]. Like other twintrons, that in the prospero gene provides a unique mechanism of alternative splicing. Remarkably, the switching between U2-dependent and U12-dependent spliceosomes is highly regulated during development and has significant consequences on the function of the gene product. More specifically, U12-dependent splicing of the twintron produces a shorter mRNA that goes on to be translated into a transcription factor with a specific homeodomain for proper DNA binding, whereas the mutually exclusive U2-dependent splicing results in a longer mRNA, that when translated, encodes a different homeodomain with potentially altered DNA-binding specificities [52]. Assuming that spliceosomal introns invaded nuclear genomes, twintrons suggest that U12 introns may not have arisen after U2 introns [48].
Furthermore, while Basu et al.’s findings were based on evidence that U12 introns positions are more conserved than those of U2 introns in humans and Arabidopsis thaliana [51], Moyer et al.’s data show that conservation in both pathways is similar, as 9% of U12 introns and 22% of U2 introns have been shown to have conserved position between humans and Arabidopsis thaliana [25]. These data argue against a parasitic invasion model in which U2 intron invasion preceded U12 invasion. On the other hand, the codivergence model assumes that following snRNA duplication in a primordial organism, introns, snRNA, and spliceosomal protein components diverged into two pathways [19,48]. Lastly, the fission/fusion model stipulates that following speciation of two different lineages from a primordial organism, the two splicing pathways diverged with each lineage containing one of the systems [19]. These organisms eventually merged, allowing the mixing of their genetic material [19]. Overall, current models support the idea that U12 and U2 introns and their splicing systems emerged around the same time.
Phylogenic reconstructions of the minor and major spliceosome’s snRNAs and proteins show that both spliceosomes existed in the last eukaryotic common ancestor (LECA) [28]. The phylogenic distribution of U12 introns includes plants, vertebrates, insects, Cnidarians, protists, and fungi [19,27]. Nonetheless, they are absent in some fungi (Saccharomyces cerevisiae and Schizosaccharomyces pombe), some protists, and nematode (Caenorhabditis elegans) [19]. The distribution of minor spliceosomal components shows the existence of a minor spliceosomal pathway in certain organisms that are within or below eukaryotic groups, suggesting loss of the minor splicing system during evolution [27]. One example is in fungi (Ascomycetes and Basidiomycetes) which lack minor spliceosomal components as opposed to Rhizopus (Zygomycota) which contains proteins components of the minor spliceosome and U12 introns. Consequently, evolutional diversification within eukaryotes may have led to an eradication of U12 introns and the minor spliceosomal system in some organisms [27]. This loss is explained by the “class conversion hypothesis”, which is believed to have occurred through the unidirectional U12 to U2 conversion. This conversion seems to preferentially occur in the GU-AG subtype of U12 introns and is unidirectional (U12 to U2) due to the constraints in the characteristics of U12 intron splice sites sequences [20,25,27]. However, it is important to note that U12 intron loss, albeit slow in vertebrate, seems to be more common than conversion [43]. A comparison of miGs across different organisms suggests that some U12 introns have been replaced by U2 introns, potentially through splice site mutations. For example, while in humans, intron 1 of PTEN is a U12 intron; in C. elegans, intron 1 is a U2 intron. Recently, a phase 0 surplus in U2 introns and an underrepresentation of phase 0 in U12 introns [25] suggested that class conversion has occurred selectively in phase 0 U12 introns with GU-AG termini, supporting the idea that class conversion (U12 to U2) occurs through subtype switching (AU-AC → GU-AG) [19]. This conversion and loss contributed to the phase biases and low abundance of U12 introns in modern eukaryotic genomes as opposed to their ancestral counterpart.
The evolutionary loss of U12 introns poses a challenge to their intrinsic feature of conservation (phylogenetically and within gene families). This intriguing discrepancy has been partially resolved with new studies suggesting that mi-INTs function is the main driver of conservation in this class of introns.
5. The Functional Relevance of Minor Introns
Minor-intron-containing genes have been described as “information processing” as opposed to “operational genes” involved in metabolism, for example [19,22,23]. mi-INTs are enriched in gene families that are involved in DNA replication/repair, transcription, translation, and RNA processing. Other enriched functional groups for miGs include voltage-gated Na and Ca channels, vesicular transport, and cytoskeletal organization [21]. Interestingly, many mi-INTs have been shown to have slow splicing kinetics (~2 fold slower than U2 introns), both in vivo [24,53] and in vitro [13,54], leading to increased mi-INT retention, while all other U2 introns in the same transcript are spliced out efficiently [24]. As the majority of miGs contain a single minor intron per gene (only a small fraction contain two or more) [55], this one intron could be rate-limiting for the expression of the miG. Thus, the nonrandom genomic distribution of mi-INTs, the fact that the majority of them exist as a single intron within miGs, and their inefficient splicing/retention led to speculations that mi-INTs play important regulatory functions. Consequently, mi-INTs were dubbed “post-transcriptional bottlenecks” which would serve to prevent gene overexpression that may be harmful to cells [24]. This is supported by the molecular switches theory [34] as shown in Figure 2A, which stipulates that mi-INTs retention forms a pool of pre-mRNA transcripts that can be rapidly expressed when needed by the cell [34]. Therefore, rather than being breaks, halting gene expression to prevent toxicity, they are considered molecular switches that can be quickly turned on or off to allow or prevent gene expression [34]. This idea was built on evidence of the high instability of U6atac snRNP, a crucial component of the minor spliceosome catalytic core [34]. This instability causes low levels of U6atac in cells leading to low levels of properly spliced mRNA for hundreds of miGs. However, once stabilized, higher U6atac rapidly enhances mi-INTs splicing and miGs expression [34]. One example of this “on-demand” miG expression is shown when U6atac stabilizes by the stress-activated protein kinase, p38MAPK, which activates these molecular switches to allow the expression of genes required to deal with cellular stress [34]. This provides an efficient mechanism through which one intron could regulate the expression of an entire transcript, a rational idea that may explain the conservation of the minor intron splicing system.
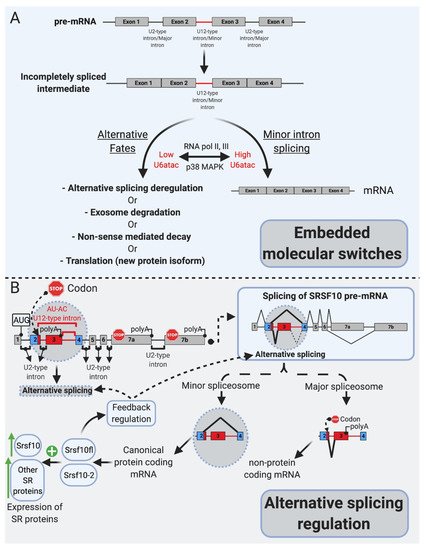
Figure 2. mi-INTs physiologic roles. (A). mi-INTs function as embedded molecular switches of gene expression [25]. Instability of U6atac result in low U6atac levels, which leads to pre-mRNA transcripts with a retained mi-INT whose fates are depicted in Figure 3. Decreased transcription lowers U6atac levels and the expression of minor-intron-containing genes, while activated p38MAPK stabilizes U6atac, increasing its levels and allowing the proper splicing of mi-INTs, producing a full-length mRNA. (B). mi-INTs regulate alternative splicing through Srsf10 [24]. Low minor spliceosome activity causes an increase in the expression of non-protein-coding Srsf10 transcript through exon 3 inclusion as a consequence of major intron splicing. High minor spliceosome activity allows exclusion of exon 3, through splicing to exon 4, allowing formation of a protein-coding transcript. The functional Srsf10 autoregulates its expression and that of other SR proteins which regulate alternative splicing of thousands of downstream targets.
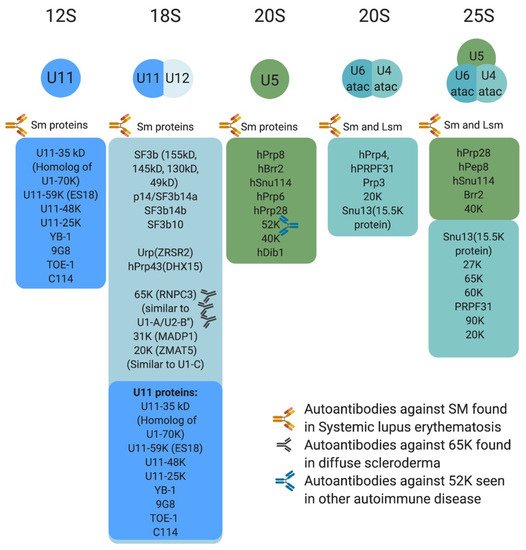
Figure 3. Protein composition of minor snRNPs and autoimmune conditions associated with them. Sm proteins include B/B’, D3, D2, D1, E, F, and G, whereas LSm proteins are LSm 2–8. The U4atac/U6atac.U5 tri-snRNP has two sets of Sm protein and one set of LSm proteins. Antibodies against some protein components of the minor spliceosome are found in systemic lupus erythematosus, diffuse scleroderma, and other autoimmune conditions.
More recent work has revealed another facet of mi-INTs as alternative splicing regulators, through the SR protein Srsf10, which itself functions in regulating alternative splicing [35]. One notable feature of the protein Srsf10 is that it autoregulates its expression through a cis regulatory element in its pre-mRNA [35] (Figure 2B). This autoregulation determines the use of competing major and minor splice sites leading to nonproductive or productive transcripts, respectively [35]. However, minor spliceosomal activity seems to partially overrule this autoregulation, as lower minor spliceosomal levels significantly lower the expression of productive Srsf10 transcripts [35], suggesting that minor spliceosomal abundance controls Srsf10 gene expression by regulating its alternative splicing [35] (Figure 2B). Furthermore, in vivo evidence suggested that the expression of many other SR proteins, which themselves do not contain mi-INTs, correlates with minor spliceosomal levels in 25 mice tissues. CRISPR/Cas9 deletion of the nonproductive Srsf10 transcript led to increased expression of many other SRSF proteins [35], establishing that minor spliceosome abundance/activity controls Srsf10 expression which in turn changes the expression of other SR proteins [35]. Taken together, these data suggest that the purpose of the minor splicing pathway is to regulate gene expression at the alternative splicing step of both miGs as well as U2 intron albeit indirectly [24,34,35].
6. mi-INTs Splicing Is Regulated
Minor-intron-containing genes show differential expression across tissues. Olthof et al. showed that both the number of miGs and their expression levels deferred across 11 mice tissues and 8 human tissues, with the heart and liver, for example expressing the least number of miGs at the lowest levels [56]. Importantly, miGs expression patterns across mice and human tissues were generally conserved [56]. Furthermore, miGs expression signatures showed functional enrichment in a tissue-specific manner [56]. For example, the cerebrum showed higher expression of miGs enriched in voltage-gated ion channel activity [56]. Intriguingly, while earlier data on a few mi-INTs have suggested an overall inefficient minor splicing pathway, recent data from 582 mi-INTs in 11 mice tissues point toward a dynamic regulatory mechanism in tissues as the inefficiency may not apply to all miGs in all tissues [56]. Therefore, mi-INTs splicing seems to be regulated in a tissue-specific manner to allow differential miGs expression. This would allow enrichment in functions that are important for specific tissues [56].
While mi-INTs retention could be an opportunity to generate a pool of unspliced transcripts standing by to be induced, it could also lead to one of the following predictable consequences (Figure 2A): (1) degradation of the host transcript by NMD or the exosome; (2) translation into a truncated protein if the transcript manages to be exported to the cytoplasm and is accessible to the ribosome, as most mi-INTs are expected to have in frame stop codons; and (3) inducing alternative splicing (AS) using cryptic or alternate splice donor, splice acceptor, or both. While the latter consequence is the least studied [21], early studies on twintrons that could switch between minor and major splicing pathways in Drosophila [52] and were later confirmed to occur in some human miGs [34], support the notion that mi-INTs provide a platform for AS. Recent work by Olthof et al. proposed that AS of mi-INTs occur more frequently than previously thought, and importantly it occurs in a tissue-specific manner [56]. Taken together, the idea that mi-INTs retention could be considered as a form of alternative splicing that is regulated agrees with the molecular switches mechanism previously described. Finally, the discovery of a high frequency of AS across this class of introns provides new avenues for the discovery of new RNA transcripts, protein isoforms/antigens that may be elevated in disease. These could therefore serve as biomarkers or targets for therapy.
7. The Minor Spliceosome: Composition, Assembly, and Evolution
The spliceosome is a multimegadalton ribonucleoprotein (RNP) complex that catalyzes the removal of introns in eukaryotes through two consecutive transesterification reactions [11,14]. The assembly of the spliceosome, which requires the recognition of intron consensus sequences at the 3′ and 5′ ends [11,14], is an organized process that involves a multitude of interactions between five U-rich small nuclear ribonucleoproteins (U snRNPs) and many associated proteins [11,14]. Upon assembly, RNA–RNA, RNA–protein, and protein–protein interactions allow for the catalysis of the splicing reaction; releasing the intron lariat and ligating the exons [11,14].
As described above, most eukaryotes contain two spliceosomes: the major (U2-dependent) spliceosome (reviewed in [11,57]), and the minor (U12-dependent) spliceosome which splices out mi-INTs. The minor spliceosome is made up of the minor snRNPs: U11, U12, U4atac, and U6atac, which are 100-fold less abundant [26,58] than their major spliceosome homologues U1, U2, U4, and U6, whereas U5 snRNP is shared by both spliceosomes. While the snRNPs composition in the two spliceosomes is homologous, there seems to be little sequence conservation between the minor and major snRNAs [50,58]. U11 and U12 sequences are unrelated to those of U1 and U2, respectively [26], while U4atac, U6atac have 40% sequence similarity to human U4 and U6, respectively [58]. Most of these conserved sequences are in regions important for RNA–RNA interaction within the spliceosome (between U6atac and U4atac or U12) or with the pre-mRNA [58]. However, some of them share analogous secondary structures, such as U4 and U6, and U4atac and U6atac interact through a base pairing interaction forming a di-snRNP secondary structure [58]. Furthermore, the U4atac/U6atac di-snRNP interacts with U5 to form a tri-snRNP similar to U4/U6.U5 tri-snRNP [50]. Of note, U1 and U2 exist as single particles that interact with RNA individually in the nucleus, and U11 and U12 form a U11/U12 di-snRNP, potentially through a protein-mediated interaction and interact with the pre-mRNA as a complex [59,60].
Protein composition of the minor spliceosome is yet another important aspect of the pathway that has received attention, leading to the identification of proteins that are unique to the minor spliceosome or shared with the major spliceosome. More than 300 protein have been identified in snRNP complexes, including snRNP-associated proteins (Sm proteins, Sm-like proteins, and snRNP-specific peptides) (Figure 3) and non-snRNP associated proteins (splicing factors) [14]. Interestingly, many of the snRNP-associated (i.e., Sm protein family) and non-snRNP-associated proteins (i.e., SR protein family) are shared between the minor and major spliceosomes [11,14,50,61]. For instance, U11/U12 di-snRNP has been shown to share all seven SF3b proteins with U2 snRNP, in addition to p14, hPrp43, YB-1 and Urp [14,21,62,63,64,65]. However, U11/U12 di-snRNP lacks all U1-specific proteins and instead contains unique proteins (65 K, 59 K, 48 K, 35 K, 31 K, 25 K, and 20 K), some of which are U11 specific [63]. This indicated that protein–protein and protein–RNA interactions at the 5′ ss are not conserved between the two spliceosomes [63]. Interestingly, these unique U11/U12 proteins are conserved in animals and plants [63,64,65]. On the other hand, the protein composition of U4atac/U6atac.U5 tri-snRNP is almost identical to that of U4/U6.U5 [50]. One notable shared protein, 15.5 K, binds the 5′ stem loop of U4 and U4atac snRNA and could play a nucleation factor role, allowing similar proteins to associate with U4/U6 and U4atac/U6atac [50,66,67,68]. However, unique minor tri-snRNP peptides remain to be identified [50]. The large number of proteins shared between the two spliceosome types suggest that protein–protein and protein–RNA interactions are similar or conserved to some extent.
The assembly of the minor spliceosome is very similar to that of the major spliceosome [13,58,60,68]. This is a stepwise process that involves the formation of multiple complexes (A, B, and C) through RNA–RNA, RNA–protein and protein–protein interactions, prior to the onset of catalysis [13,48,58,60,68] (Figure 1B). Early assembly involves the recognition of the 5′ ss and BPS cooperatively, through U11/5′ ss and U12/BPS interactions by the U11/U12 di-snRNP, which allows the formation of pre-spliceosomal complex A [13,58,60]. Subsequently, snRNA–snRNA base pairing allows the formation of U4atac/U6atac di-snRNP in which U4atac functions as an RNA chaperone to allow 5′ ss recognition by U6atac [48,58]. U4atac/U6atac and U5 are recruited to the spliceosome to form mature spliceosomal complex B [13,58]. The U4atac/U6atac.U5 tri-snRNP interaction disrupts the base pairing between U4atac/U6atac displacing U4atac before the first catalytic reaction [48,58]. U5 interacts with the exonic sequences at the 5′ ss and 3′ ss, and U6atac base pairs with the 5′ ss displacing U11 and interacting with U12 [48,58]. Consequently, U6atac and U12 form the activated catalytic core while U5 aligns the two exons for the second catalytic step, these form complex C that is catalytically active [13,48,58,68].
The identification of the snRNA and protein components in the minor spliceosome has helped shed light on the evolutionary relationship between the minor and major splicing systems. This relationship was described by the three models (previously discussed), which attempt to answer the question of whether these two systems originated from a common ancestor (homologous origin) [19]. The fact that most proteins in U4/U6.U5 are shared with U4atac/U6atac.U5, while many other proteins are shared between U2 and U11/U12 [50,61,65], supports a model of common ancestry [19]. However, the identification of novel unique U11/U12 proteins that have 5′ ss interactions, distinct from U1 [63], challenges the homologous origin model. This is further exacerbated by evidence of low percentage/lack of sequence conservation between minor and major spliceosomal snRNAs [26,58]. Therefore, a nonhomologous model such as a parasitic group II intron invasion model could explain the highly diverged snRNA and the high protein similarity in the minor and major spliceosomes [19,50,63]. However, this model does not explain the novel proteins identified in the minor spliceosome [63]. Instead, a homologous model with high divergence such as the fission fusion model could account for the current observations [19,63]. Speciation may have led to two separate lineages, each containing one splicing pathway in which snRNAs and spliceosomal proteins diverged separately [19,63]. Eventually, endosymbiosis allowed the fusion of the two lineages, forming an ancestral eukaryote with both splicing systems [19,63].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22116062
This entry is offline, you can click here to edit this entry!