Dietary polyphenols are a group of natural compounds that have been proposed to have beneficial effects on human health. They were first known for their antioxidant properties, but several studies over the years have shown that these compounds can exert protective effects against chronic diseases. Nonetheless, the mechanisms underlying these potential benefits are still uncertain and contradictory effects have been reported. The effect of these compounds on visual health, and particularly on retinal degenerative diseases, is a matter of renewed interest and recent studies show promising results for the use of these compounds to improve visual function.
- flavonoids
- retinal degenerative diseases
- retinitis pigmentosa
- protein folding
- ligand binding
- rhodopsin
1. Introduction
2. Polyphenols as Repurposed Drugs
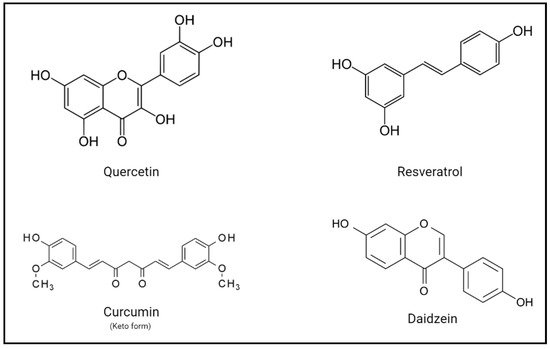
3. Implications and Potential Benefits of Polyphenols on Human Health
4. Vertebrate Rho and Retinal Degeneration
4.1. Rho as a GPCR
4.2. Visual Phototransduction
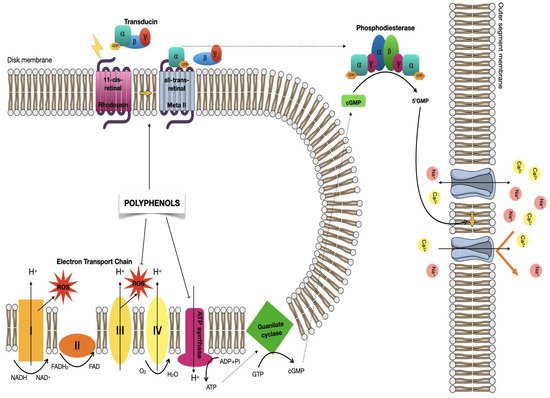
4.3. Mutations in Rho Associated with Retinal Degenerative Diseases
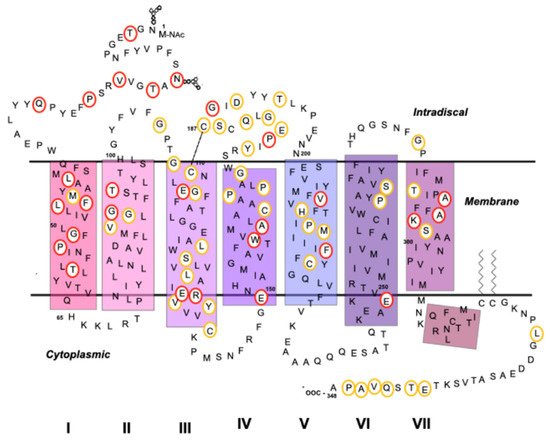
| Mutation | Behavior/Effect | Class/Misfolds | References |
|---|---|---|---|
| G90X | Causes thermal instability and/or abnormal photoproduct formation in inducing a RP phenotype. | VI/No | [45] |
| T94I | Induces constitutive activation of the opsin in the absence of chromophore and in the dark. | VI/No | [94] |
| E113K | Associated with the two distinct phenotypes of RP and CSNB in independent members of the same family. | Unclassified | [91] |
| A292E | Anomalously activates transducin when the chromophore is missing. | Unclassified | [92] |
| P23H | Destabilizes outer rod segments via the formation of aggregates due to retention in the ER. | II/Yes | [87] |
| E150 | No observed biochemical or cellular defects or not studied in detail. | Unclassified | [101] |
| W161X | No observed biochemical or cellular defects or not studied in detail. | Unclassified | [103] |
| G114V | No observed biochemical or cellular defects or not studied in detail. | Unclassified | [107] |
| Q184P | No observed biochemical or cellular defects or not studied in detail. | Unclassified | [107] |
| R135X | Affects endocytosis | III/No | [108] |
| G188R | Forms aggregates due to retention in the ER and cannot be easily constituted with 11CR. | II/Yes | [109] |
5. Polyphenols Effects in Retinal Degenerative Diseases
Therapies for retinal degenerative diseases are currently limited, so there is a need to develop new strategies for more effective and safer therapies. As we have seen, polyphenols, especially flavonoids, could be viable drug candidates as they may be involved in visual signal transduction and visual pigment regeneration. Flavonoid-rich vegetables and fruits appear to have effects in improving eyesight in eye-related diseases[61][122][123].
We will focus on the effects of flavonoids in three different retinal-related diseases: RP, CSNB and age-related macular degeneration (AMD).
RP has already been described previously. Additionally, CSNB is a group of heterogeneous genetic disorders of the retina that manifest as non-progressive nyctalopia [124]. Finally, AMD is a complex disease that exhibits several different pathological mechanisms including degeneration of photoreceptors and RPE cells causing visual impairment [125].
Flavonoids such as quercetin and myricetin have been shown to improve the stability of opsin present in rods, increase the binding rate of ligand-free opsin, and facilitate its expression and integration into the membrane in vitro [126]. In spite of the studies presenting beneficial effects of flavonoids, the mechanisms of their protective effects against light-induced retinal damage are not entirely known [123]. Some studies suggest that flavonoids interact directly with Rho, increasing their rates of regeneration, stability, folding, and membrane orientation in vitro and have an effect on retinal degenerative diseases (Table 2) [46][127][128].
Table 2. Summary of different polyphenols effects on retinal physiology.
|
Compound |
Condition/Cell Lines |
Effect |
References |
|
Quercetin |
Oxidative stress conditions. Assay in vitro in human hepatoma HepG2 cells. |
Activates the Nrf2-ARE signaling pathway and exhibits anti-oxidative stress activity alone and together with kaempferol and pterostilbene. |
[123] |
|
Oxidative stress conditions. Assay in vitro in human RPE cells and in Ccl2/Cx3cr1 double knock-out mice. |
Protects RPE cells from oxidative stress via inhibiting pro-inflammatory molecules and the intrinsic apoptosis pathway. |
[129] |
|
|
VEGF-treated mouse photoreceptor-derived 661W cells. |
Inhibits the production of inflammatory proteins in VEGF-stimulated 661W cells. |
[130] |
|
|
Oxidative stress conditions. ARPE-19 human retinal pigment epithelial cells. |
Protects ARPE-19 cells from H2O2-induced cytotoxicity by activating the Nrf2 pathway, inhibiting ER stress and targeting anti-apoptotic proteins. |
[131] |
|
|
Oxidative stress conditions. Assay in vitro in human RPE cells. |
Protects RPE cells from oxidative damage and cellular senescence in a dose-dependent manner. |
[132] |
|
|
Oxidative stress conditions. Assay in vitro and in vivo in human RPE cells. |
Protects against blue light-induced retinal damage. |
[133] |
|
|
Myricetin |
Human MCF-7 breast cancer cells. |
Reduces and scavenges intracellular ROS. |
[134] |
|
Apigenin |
Bright light-exposed BALB/c mice. |
Confers retinal protection by inhibiting retinal oxidative stress and retinal inflammatory responses. |
[135] |
|
Tannic acid |
Assay in vitro in human RPE cells (ARPE-19). |
Protects RPE against ultraviolet B radiation via the inhibition of the inflammatory response. |
[136] |
|
Fisetin/Luteolin |
Assay in vitro in human RPE cells (ARPE-19). |
Anti-inflammatory and cytoprotective effects when used as dietary supplements. |
[137] |
Flavonoids have been found to stimulate Rho expression, where specifically Rho and cone opsins expression have been improved upon treatment with quercetin and myricetin [127][138][139].
The antioxidant effect of polyphenols can be invoked as a factor that may delay the progression of AMD. A particular compound, namely, stilbenoid resveratrol, a dietary compound with a wide range of effects on cell function, has been shown to effectively reduce ROS production, thus protecting against retinal damage [84][140].
Flavonoids can also inhibit inflammatory reactions by suppressing the expression of pro-inflammatory genes and molecules involved in retinal degeneration. In addition, they can also limit ROS levels by sequestering oxidative radicals. In this regard, RPE cells treated with quercetin could be protected from oxidative stress by inhibiting apoptosis pathways and pro-inflammatory markers [134][141]. Flavonoids enhance the expression of photoreceptor-specific genes by also attenuating the expression of oxidative stress and inflammation-related malignancies and altering the balance between anti-apoptotic and pro-apoptotic genes [129][130][131][135].
Another polyphenol, tannic acid, has also been reported to inhibit the production of interleukin-6 and to down-regulate the expression of complement factor B in ARPE-19 cells, a factor that is believed to be related to AMD [136].
In ARPE-19 cells, quercetin protects against stress induced by lipid peroxidation [142]. Quercetin was observed to reduce mitochondrial function protecting against hydrogen peroxide-induced oxidative stress in RPE cells of human donor eyes thus increasing its viability [132]. Other studies have shown that this polyphenol can improve oxidative stress and its consequences in different regions of the eye [133][143][144][145].
Quercetin has also produced a protective effect against oxidative stress and its consequences on photoreceptor cells resulting from the reaction of ATR with phosphatidylethanolamine producing bis-retinoid photoreactive species [146].
Mechanisms involved in the antioxidant activity of polyphenols include suppression of ROS formation [147][148], thus reducing oxidative damage [149]. The mechanism by which ROS formation is reduced involves phosphorylation of Nrf2 residues resulting in nuclear accumulation [150].
Although the implication of flavonoids in vision and vision diseases is still uncertain, some studies with dietary flavonoids like quercetin have suggested potential beneficial effects in some forms of RP [45]. Mutations in Rho are associated with this disease and they can cause protein misfolding that leads to a progressive loss of rod and cone cells, further resulting in vision loss [151][152][153]. These results should be analyzed in the context of research in the RP field, where several strategies based on pharmacological rescue have been proposed for RP treatment. The basic principle of this approach is that chemical or pharmacological chaperones bind to misfolded opsins and are able to stabilize them [154].
The dietary flavonoid quercetin, one of the most studied and widely known for its potential beneficial effects on health [155], has been used in some experiments with the recombinant G90V mutant associated with RP and has shown satisfactory effects when combined with 9-cis-retinal (9CR), a retinal analog that is usually employed in vision studies. Over the past years, different investigations have focused on describing the pharmaceutical application of 9-cis retinoids to remedy the retinal dysfunction caused by deficient regeneration with 11CR [156][157][158] and have shown that this retinal analog can increase the stability of the RP mutant G90V [70].
In summary, the use of polyphenols, like quercetin, alone or in combination with other small ligands, like retinoids, opens new possibilities for the treatment of retinal degeneration associated with RP. Moreover, the new effect attributed to quercetin may also be applicable to other members of the GPCR superfamily [45]. In spite of these encouraging results, there is clearly a need to further investigate the in vivo potential of such strategies and particularly to increase the number of clinical studies being performed. This is essential to fully determine the exact reach of these newly proposed mechanisms and the potential physiological effects of specific compounds.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26113407
References
- Rasouli, H.; Farzaei, M.H.; Khodarahmi, R. Polyphenols and their benefits: A review. Int. J. Food Prop. 2017, 20, 1700–1741.
- Xiao, J.B. Stability of dietary polyphenols: It’s never too late to mend? Food Chem. Toxicol. 2018, 119, 3–5.
- Sies, H. Polyphenols and health: Update and perspectives. Arch. Biochem. Biophys. 2010, 501, 2–5.
- World Health Organization. Diet, Nutrition and the Prevention of Chronic Diseases; World Health Organization: Geneva, Switzerland, 2003.
- Hollman, P.C.; Geelen, A.; Kromhout, D. Dietary flavonol intake may lower stroke risk in men and women. J. Nutr. 2010, 140, 600–604.
- Hooper, L.; Kroon, P.A.; Rimm, E.B.; Cohn, J.S.; Harvey, I.; Le Cornu, K.A.; Ryder, J.J.; Hall, W.L.; Cassidy, A. Flavonoids, flavonoid-rich foods, and cardiovascular risk: A meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2008, 88, 38–50.
- Lupton, J.R.; Atkinson, S.A.; Chang, N.; Fraga, C.G.; Levy, J.; Messina, M.; Richardson, D.P.; van Ommen, B.; Yang, Y.; Griffiths, J.C.; et al. Exploring the benefits and challenges of establishing a DRI-like process for bioactives. Eur. J. Nutr. 2014, 53, 1–9.
- Kennedy, D.O. Polyphenols and the human brain: Plant “secondary metabolite” ecologic roles and endogenous signaling functions drive benefits. Adv. Nutr. 2014, 5, 515–533.
- Marin, L.; Miguelez, E.M.; Villar, C.J.; Lombo, F. Bioavailability of dietary polyphenols and gut microbiota metabolism: Antimicrobial properties. Biomed. Res. Int. 2015, 2015, 1–18.
- Tsao, R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246.
- Miyata, Y.; Shida, Y.; Hakariya, T.; Sakai, H. Anti-cancer effects of green tea polyphenols against prostate cancer. Molecules 2019, 24, 193.
- Serino, A.; Salazar, G. Protective role of polyphenols against vascular inflammation, aging and cardiovascular disease. Nutrients 2018, 11, 53.
- Leri, M.; Scuto, M.; Ontario, M.L.; Calabrese, V.; Calabrese, E.J.; Bucciantini, M.; Stefani, M. Healthy effects of plant polyphenols: Molecular mechanisms. Int. J. Mol. Sci. 2020, 21, 1250.
- Cho, E.; Seddon, J.M.; Rosner, B.; Willett, W.C.; Hankinson, S.E. Prospective study of intake of fruits, vegetables, vitamins, and carotenoids and risk of age-related maculopathy. Arch. Ophthalmol. 2004, 122, 883.
- Tang, L.; Zhang, Y.; Jiang, Y.; Willard, L.; Ortiz, E.; Wark, L.; Medeiros, D.; Lin, D. Dietary wolfberry ameliorates retinal structure abnormalities in db/db mice at the early stage of diabetes. Exp. Biol. Med. 2011, 236, 1051–1063.
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S.
- Cheynier, V. Polyphenols in foods are more complex than often thought. Am. J. Clin. Nutr. 2005, 81, 223S–229S.
- Wen, L.; Jiang, Y.; Yang, J.; Zhao, Y.; Tian, M.; Yang, B. Structure, bioactivity, and synthesis of methylated flavonoids. Ann. N. Y. Acad. Sci. 2017, 1398, 120–129.
- Selma, M.V.; Espín, J.C.; Tomás-Barberán, F.A. Interaction between phenolics and gut microbiota: Role in human health. J. Agric. Food Chem. 2009, 57, 6485–6501.
- Crozier, A.; Del Rio, D.; Clifford, M.N. Bioavailability of dietary flavonoids and phenolic compounds. Mol. Aspects. Med. 2010, 31, 446–467.
- Galleano, M.; Verstraeten, S.V.; Oteiza, P.I.; Fraga, C.G. Antioxidant actions of flavonoids: Thermodynamic and kinetic analysis. Arch. Biochem. Biophys. 2010, 501, 23–30.
- Fraga, C.G. Plant polyphenols: How to translate their in vitro antioxidant actions to in vivo conditions. IUBMB Life 2007, 59, 308–315.
- Focaccetti, C.; Izzi, V.; Benvenuto, M.; Fazi, S.; Ciuffa, S.; Giganti, M.G.; Potenza, V.; Manzari, V.; Modesti, A.; Bei, R. Polyphenols as immunomodulatory compounds in the tumor microenvironment: Friends or foes? Int. J. Mol. Sci. 2019, 20, 1714.
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124.
- Dudnik, A.; Gaspar, P.; Neves, A.R.; Forster, J. Engineering of microbial cell factories for the production of plant polyphenols with health-beneficial properties. Curr. Pharm. Des. 2018, 24, 2208–2225.
- Fraga, C.G.; Oteiza, P.I.; Galleano, M. Plant bioactives and redox signaling: (–)-Epicatechin as a paradigm. Mol. Aspects. Med. 2018, 61, 31–40.
- Kim, H.-S.; Quon, M.J.; Kim, J. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014, 2, 187–195.
- Fraga, C.G.; Croft, K.D.; Kennedy, D.O.; Tomás-Barberán, F.A. The effects of polyphenols and other bioactives on human health. Food Funct. 2019, 10, 514–528.
- Cremonini, E.; Bettaieb, A.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. (–)-Epicatechin improves insulin sensitivity in high fat diet-fed mice. Arch. Biochem. Biophys. 2016, 599, 13–21.
- Vazquez-Prieto, M.A.; Bettaieb, A.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. (−)-Epicatechin prevents TNFα-induced activation of signaling cascades involved in inflammation and insulin sensitivity in 3T3-L1 adipocytes. Arch. Biochem. Biophys. 2012, 527, 113–118.
- Bettaieb, A.; Cremonini, E.; Kang, H.; Kang, J.; Haj, F.G.; Oteiza, P.I. Anti-inflammatory actions of (−)-epicatechin in the adipose tissue of obese mice. Int. J. Biochem. Cell Biol. 2016, 81, 383–392.
- Crichton, G.E.; Elias, M.F.; Dearborn, P.; Robbins, M. Habitual chocolate intake and type 2 diabetes mellitus in the Maine-Syracuse Longitudinal Study: (1975–2010): Prospective observations. Appetite 2017, 108, 263–269.
- Buitrago-Lopez, A.; Sanderson, J.; Johnson, L.; Warnakula, S.; Wood, A.; Di Angelantonio, E.; Franco, O.H. Chocolate consumption and cardiometabolic disorders: Systematic review and meta-analysis. BMJ 2011, 343, d4488.
- Khan, N.; Mukhtar, H. Tea polyphenols in promotion of human health. Nutrients 2018, 11, 39.
- Bondonno, N.P.; Bondonno, C.P.; Blekkenhorst, L.C.; Considine, M.J.; Maghzal, G.; Stocker, R.; Woodman, R.J.; Ward, N.C.; Hodgson, J.M.; Croft, K.D. Flavonoid-rich apple improves endothelial function in individuals at risk for cardiovascular disease: A randomized controlled clinical trial. Mol. Nutr. Food Res. 2017, 62, 1700674.
- Huang, H.; Chen, G.; Liao, D.; Zhu, Y.; Xue, X. Effects of berries consumption on cardiovascular risk factors: A Meta-analysis with trial sequential analysis of randomized controlled trials. Sci. Rep. 2016, 6, 23625.
- Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Interactions of gut microbiota with dietary polyphenols and consequences to human health. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 471–476.
- Oteiza, P.I.; Fraga, C.G.; Mills, D.A.; Taft, D.H. Flavonoids and the gastrointestinal tract: Local and systemic effects. Mol. Aspects Med. 2018, 61, 41–49.
- Crichton, G.E.; Elias, M.F.; Alkerwi, A. Chocolate intake is associated with better cognitive function: The Maine-Syracuse Longitudinal Study. Appetite 2016, 100, 126–132.
- Moreira, A.; Diógenes, M.J.; de Mendonça, A.; Lunet, N.; Barros, H. Chocolate consumption is associated with a lower risk of cognitive decline. J. Alzheimers Dis. 2016, 53, 85–93.
- Ng, T.P.; Feng, L.; Niti, M.; Kua, E.H.; Yap, K.B. Tea consumption and cognitive impairment and decline in older Chinese adults. Am. J. Clin. Nutr. 2008, 88, 224–231.
- Kuriyama, S.; Hozawa, A.; Ohmori, K.; Shimazu, T.; Matsui, T.; Ebihara, S.; Awata, S.; Nagatomi, R.; Arai, H.; Tsuji, I. Green tea consumption and cognitive function: A cross-sectional study from the Tsurugaya Project. Am. J. Clin. Nutr. 2006, 83, 355–361.
- Dong, X.; Yang, C.; Cao, S.; Gan, Y.; Sun, H.; Gong, Y.; Yang, H.; Yin, X.; Lu, Z. Tea consumption and the risk of depression: A meta-analysis of observational studies. Aust. N. Z. J. Psychiatry 2015, 49, 334–345.
- Li, F.-J.; Ji, H.-F.; Shen, L. A Meta-analysis of tea drinking and risk of Parkinson’s disease. Sci. World J. 2012, 2012, 1–6.
- Herrera-Hernández, M.G.; Ramon, E.; Lupala, C.S.; Tena-Campos, M.; Pérez, J.J.; Garriga, P. Flavonoid allosteric modulation of mutated visual rhodopsin associated with retinitis pigmentosa. Sci. Rep. 2017, 7, 11167.
- Bourne, H.R.; Meng, E.C. Structure. Rhodopsin sees the light. Science 2000, 289, 733–734.
- Nickell, S.; Park, P.S.-H.; Baumeister, W.; Palczewski, K. Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J. Cell Biol. 2007, 177, 917–925.
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745.
- Jastrzebska, B. GPCR: G protein complexes—The fundamental signaling assembly. Amino Acids 2013, 45, 1303–1314.
- Katayama, K.; Gulati, S.; Ortega, J.T.; Alexander, N.S.; Sun, W.; Shenouda, M.M.; Palczewski, K.; Jastrzebska, B. Specificity of the chromophore-binding site in human cone opsins. J. Biol. Chem. 2019, 294, 6082–6093.
- Zhang, D.; Zhao, Q.; Wu, B. Structural studies of G protein-coupled receptors. Mol. Cells 2015, 38, 836–842.
- Alexander, S.P.H.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; Spedding, M.; Peters, J.A.; Harmar, A.J. The concise guide to PHARMACOLOGY 2013/14: G proteincoupled receptors. Br. J. Pharmacol. 2013, 170, 1459–1581.
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272.
- Stevens, R.C.; Cherezov, V.; Katritch, V.; Abagyan, R.; Kuhn, P.; Rosen, H.; Wüthrich, K. The GPCR network: A large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov. 2013, 12, 25–34.
- Lindsley, C.W.; Emmitte, K.A.; Hopkins, C.R.; Bridges, T.M.; Gregory, K.J.; Niswender, C.M.; Conn, P.J. Practical strategies and concepts in GPCR allosteric modulator discovery: Recent advances with metabotropic glutamate receptors. Chem. Rev. 2016, 116, 6707–6741.
- Khoury, E.; Clément, S.; Laporte, S.A. Allosteric and biased g protein-coupled receptor signaling regulation: Potentials for new therapeutics. Front. Endocrinol. 2014, 5, 68.
- Sato, J.; Makita, N.; Iiri, T. Inverse agonism: The classic concept of GPCRs revisited. Endocr. J. 2016, 63, 507–514.
- Lane, J.R.; Abdul-Ridha, A.; Canals, M. Regulation of G protein-coupled receptors by allosteric ligands. ACS Chem. Neurosci. 2013, 4, 527–534.
- Hubbard, R.; Kropf, A. The action of light on rhodopsin. Proc. Natl. Acad. Sci. USA 1958, 44, 130–139.
- Nakamichi, H.; Okada, T. X-ray crystallographic analysis of 9-cis-rhodopsin, a model analogue visual pigment. J. Photochem. Photobiol. 2007, 83, 232–235.
- Kalt, W.; Hanneken, A.; Milbury, P.; Tremblay, F. Recent research on polyphenolics in vision and eye health. J. Agric. Food Chem. 2010, 58, 4001–4007.
- Zhong, M.; Kawaguchi, R.; Kassai, M.; Sun, H. Retina, retinol, retinal and the natural history of vitamin A as a light sensor. Nutrients 2012, 4, 2069–2096.
- Park, J.H.; Scheerer, P.; Hofmann, K.P.; Choe, H.-W.; Ernst, O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 2008, 454, 183–187.
- Garriga, P.; Manyosa, J. The eye photoreceptor protein rhodopsin. Structural implications for retinal disease. FEBS Lett. 2002, 528, 17–22.
- Ridge, K.D.; Abdulaev, N.G.; Sousa, M.; Palczewski, K. Phototransduction: Crystal clear. Trends Biochem. Sci. 2003, 28, 479–487.
- Travis, G.H.; Golczak, M.; Moise, A.R.; Palczewski, K. Diseases caused by defects in the visual cycle: Retinoids as potential therapeutic agents. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 469–512.
- Calzia, D.; Barabino, S.; Bianchini, P.; Garbarino, G.; Oneto, M.; Caicci, F.; Diaspro, A.; Tacchetti, C.; Manni, L.; Candiani, S.; et al. New findings in ATP supply in rod outer segments: Insights for retinopathies. Biol. Cell. 2013, 105, 345–358.
- Kiser, P.D.; Golczak, M.; Palczewski, K. Chemistry of the Retinoid (Visual) Cycle. Chem. Rev. 2014, 114, 194–232.
- Fan, J.; Woodruff, M.L.; Cilluffo, M.C.; Crouch, R.K.; Fain, G.L. Opsin activation of transduction in the rods of dark-reared Rpe65 knockout mice. J. Physiol. 2005, 568, 83–95.
- Toledo, D.; Ramon, E.; Aguilà, M.; Cordomí, A.; Pérez, J.J.; Mendes, H.F.; Cheetham, M.E.; Garriga, P. Molecular mechanisms of disease for mutations at Gly-90 in rhodopsin. J. Biol. Chem. 2011, 286, 39993–40001.
- Palczewski, K. G protein-coupled receptor rhodopsin. Annu. Rev. Biochem. 2006, 75, 743–767.
- Veleri, S.; Lazar, C.H.; Chang, B.; Sieving, P.A.; Banin, E.; Swaroop, A. Biology and therapy of inherited retinal degenerative disease: Insights from mouse models. Dis. Models Mech. 2015, 8, 109–129.
- Chen, Y.; Okano, K.; Maeda, T.; Chauhan, V.; Golczak, M.; Maeda, A.; Palczewski, K. Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J. Biol. Chem. 2012, 287, 5059–5069.
- Kim, S.R.; Jang, Y.P.; Jockusch, S.; Fishkin, N.E.; Turro, N.J.; Sparrow, J.R. The all-trans- retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. Proc. Natl. Acad. Sci. USA 2007, 104, 19273–19278.
- Sparrow, J.R.; Wu, Y.; Kim, C.Y.; Zhou, J. Phospholipid meets all-trans-retinal: The making of RPE bisretinoids. J. Lipid. Res. 2010, 51, 247–261.
- Gao, S.; Parmar, T.; Palczewska, G.; Dong, Z.; Golczak, M.; Palczewski, K.; Jastrzebska, B. Protective effect of a locked retinal chromophore analog against light-induced retinal degeneration. Mol. Pharmacol. 2018, 94, 1132–1144.
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of mitochondrial DNA damage in ROS-mediated pathogenesis of Age-related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374.
- Sawada, O.; Perusek, L.; Kohno, H.; Howell, S.J.; Maeda, A.; Matsuyama, S.; Maeda, T. All-trans-retinal induces Bax activation via DNA damage to mediate retinal cell apoptosis. Exp. Eye Res. 2014, 123, 27–36.
- Kohno, H.; Maeda, T.; Perusek, L.; Pearlman, E.; Maeda, A. CCL3 production by microglial cells modulates disease severity in murine models of retinal degeneration. J. Immunol. 2014, 192, 3816–3827.
- Rashid, K.; Wolf, A.; Langmann, T. Microglia activation and immunomodulatory therapies for retinal degenerations. Front. Cell. Neurosci. 2018, 12, 176.
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975.
- Bruschi, M.; Bartolucci, M.; Peteretto, A.; Calzia, D.; Caicci, F.; Manni, L.; Traverso, C.E.; Candiano, G.; Panfoli, I. Differential expression of the five redox complexes in the retinal mitochondria or rod outer segment disks is consistent with their different functionality. FASEB BioAdv. 2020, 2, 315–324.
- Bruschi, M.; Petretto, A.; Caicci, F.; Bartolucci, M.; Calzia, D.; Santucci, L.; Manni, L.; Ramenghi, L.A.; Ghiggeri, G.; Traverso, C.E.; et al. Proteome of bovine mitochondria and rod outer segments disks: Commonalities and differences. J. Proteome Res. 2018, 17, 918–925.
- Ravera, S.; Esposito, A.; Degan, P.; Caicci, F.; Calzia, D.; Perrotta, E.; Manni, L.; Bisio, A.; Iobbi, V.; Schito, A.; et al. Sclareol modulates free radical production in the retinal rod outer segment by inhibiting the ectopic f1fo-atp synthase. Free Radic. Biol. Med. 2020, 60, 368–375.
- Goldberg, A.F.; Moritz, O.L.; Williams, D.S. Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res. 2016, 55, 52–81.
- Athanasiou, D.; Aquila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23.
- Dryja, T.P.; McGee, T.L.; Hahn, L.B.; Cowley, G.S.; Olsson, J.E.; Reichel, E.; Sandberg, M.A.; Berson, E.L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990, 323, 1302–1307.
- Rao, V.R.; Cohen, G.B.; Oprian, D.D. Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature 1994, 367, 639–642.
- Sieving, P.A.; Richards, J.E.; Naarendorp, F.; Bingham, E.L.; Scott, K.; Alpern, M. Dark-light: Model for night blindness from the human rhodopsin Gly-90-->Asp mutation. Proc. Natl. Acad. Sci. USA 1995, 92, 880–884.
- Al-Jandal, N.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Bannon, N.; Findlay, J.B.; Humphries, P.; Kenna, P.F. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum. Mutat. 1999, 13, 75–81.
- Reiff, C.; Owczarek-Lipska, M.; Spital, G.; Roger, C.; Hinz, H.; Juschke, C.; Thiele, H.; Altmuller, J.; Nurnberg, P.; Da Costa, R.; et al. The mutation p.E113K in the Schiff base counterion of rhodopsin is associated with two distinct retinal phenotypes within the same family. Sci. Rep. 2016, 6, 36208.
- Dryja, T.P.; Berson, E.L.; Rao, V.R.; Oprian, D.D. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat. Genet. 1993, 4, 280–283.
- Zeitz, C.; Gross, A.K.; Leifert, D.; Kloeckener-Gruissem, B.; McAlear, S.D.; Lemke, J.; Neidhardt, J.; Berger, W. Identification and functional characterization of a novel rhodopsin mutation associated with autosomal dominant CSNB. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4105–4114.
- Gross, A.K.; Rao, V.R.; Oprian, D.D. Characterization of rhodopsin congenital night blindness mutant T94I. Biochemistry 2003, 42, 2009–2015.
- Singhal, A.; Guo, Y.; Matkovic, M.; Schertler, G.; Deupi, X.; Yan, E.C.; Standfuss, J. Structural role of the T94I rhodopsin mutation in congenital stationary night blindness. EMBO Rep. 2016, 17, 1431–1440.
- Chen, J.; Shi, G.; Concepcion, F.A.; Xie, G.; Oprian, D.; Chen, J. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J. Neurosci. 2006, 26, 11929–11937.
- Tam, B.M.; Moritz, O.L. Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3234–3241.
- Ramon, E.; del Valle, L.J.; Garriga, P. Unusual thermal and conformational properties of the rhodopsin congenital night blindness mutant Thr-94 --> Ile. J. Biol. Chem. 2003, 278, 6427–6432.
- Azam, M.; Khan, M.I.; Gal, A.; Hussain, A.; Shah, S.T.; Khan, M.S.; Sadeque, A.; Bokhari, H.; Collin, R.W.J.; Orth, U.; et al. A homozygous p.Glu150Lys mutation in the opsin gene of two Pakistani families with autosomal recessive retinitis pigmentosa. Mol. Vis. 2009, 15, 2526–2534.
- Saqib, M.A.; Nikopoulos, K.; Ullah, E.; Sher Khan, F.; Iqbal, J.; Bibi, R.; Jarral, A.; Sajid, S.; Nishiguchi, K.M.; Venturini, G.; et al. Homozygosity mapping reveals novel and known mutations in Pakistani families with inherited retinal dystrophies. Sci. Rep. 2015, 5, 9965.
- Van Schil, K.; Karlstetter, M.; Aslanidis, A.; Dannhausen, K.; Azam, M.; Qamar, R.; Leroy, B.P.; Depasse, F.; Langmann, T.; De Baere, E. Autosomal recessive retinitis pigmentosa with homozygous rhodopsin mutation E150K and non-coding cis-regulatory variants in CRX-binding regions of SAMD7. Sci. Rep. 2016, 6, 21307.
- Collin, R.W.; van den Born, L.I.; Klevering, B.J.; de Castro-Miro, M.; Littink, K.W.; Arimadyo, K.; Azam, M.; Yazar, V.; Zonneveld, M.N.; Paun, C.C.; et al. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2227–2239.
- Kartasasmita, A.; Fujiki, K.; Iskandar, E.; Sovani, I.; Fujimaki, T.; Murakami, A. A novel nonsense mutation in rhodopsin gene in two Indonesian families with autosomal recessive retinitis pigmentosa. Ophthalmic Genet. 2011, 32, 57–63.
- Rosenfeld, P.J.; Cowley, G.S.; McGee, T.L.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat. Genet. 1992, 1, 209–213.
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3052–3064.
- Jacobson, S.G.; Kemp, C.M.; Sung, C.H.; Nathans, J. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am. J. Ophthalmol. 1991, 112, 256–271.
- Dryja, T.P.; McEvoy, J.A.; McGee, T.L.; Berson, E.L. Novel rhodopsin mutations Gly114Val and Gln184Pro in dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3124–3127.
- Shi, W.; Sports, C.D.; Raman, D.; Shirakawa, S.; Osawa, S.; Weiss, E.R. Rhodopsin arginine-135 mutants are phosphorylated by rhodopsin kinase and bind arrestin in the absence of 11-cis retinal. Biochemistry 1998, 37, 4869–4874.
- Del Porto, G.; Vingolo, E.M.; David, D.; Steindl, K.; Wedemann, H.; Forte, R.; Iannccone, A.; Gal, A.; Pannarale, M.R. Clinical features of autosomal dominant retinitis pigmentosa associated with the GLY-188-ARG mutation of the rhodopsin gene. In Retinal Degeneration; Hollyfield, J.G., Anderson, R.E., LaVail, M.M., Eds.; Springer: Boston, MA, USA, 1993; pp. 91–101.
- Van Woerkom, C.; Ferrucci, S. Sector retinitis pigmentosa. Optometry 2005, 76, 309–317.
- Ramon, E.; Cordomi, A.; Aguila, M.; Srinivasan, S.; Dong, X.; Moore, A.T.; Webster, A.R.; Cheetham, M.E.; Garriga, P. Differential light-induced responses in sectorial inherited retinal degeneration. J. Biol. Chem. 2014, 289, 35918–35928.
- Sanchez-Reyes, O.B.; Cooke, A.L.G.; Tranter, D.B.; Rashid, D.; Eilers, M.; Reeves, P.J.; Smith, S.O. G Protein-Coupled Receptors Contain Two Conserved Packing Clusters. Biophys. J. 2017, 112, 2315–2326.
- Jastrzebska, B.; Chen, Y.; Orban, T.; Jin, H.; Hofmann, L.; Palczewski, K. Disruption of rhodopsin dimerization with synthetic peptides targeting an interaction interface. J. Biol. Chem. 2015, 290, 25728–25744.
- Kota, P.; Reeves, P.J.; Rajbhandary, U.L.; Khorana, H.G. Opsin is present as dimers in COS1 cells: Identification of amino acids at the dimeric interface. Proc. Natl. Acad. Sci. USA 2006, 103, 3054–3059.
- Gunkel, M.; Schoneberg, J.; Alkhaldi, W.; Irsen, S.; Noe, F.; Kaupp, U.B.; Al-Amoudi, A. Higher-order architecture of rhodopsin in intact photoreceptors and its implication for phototransduction kinetics. Structure 2015, 23, 628–638.
- Ploier, B.; Caro, L.N.; Morizumi, T.; Pandey, K.; Pearring, J.N.; Goren, M.A.; Finnemann, S.C.; Graumann, J.; Arshavsky, V.Y.; Dittman, J.S.; et al. Dimerization deficiency of enigmatic retinitis pigmentosa-linked rhodopsin mutants. Nat. Commun. 2016, 7, 12832.
- Davies, W.I.; Downes, S.M.; Fu, J.K.; Shanks, M.E.; Copley, R.R.; Lise, S.; Ramsden, S.C.; Black, G.C.M.; Gibson, K.; Foster, R.G.; et al. Next-generation sequencing in health-care delivery: Lessons from the functional analysis of rhodopsin. Genet. Med. 2012, 14, 891–899.
- Lim, K.P.; Yip, S.P.; Cheung, S.C.; Leung, K.W.; Lam, S.T.; To, C.H. Novel PRPF31 and PRPH2 mutations and co-occurrence of PRPF31 and RHO mutations in Chinese patients with retinitis pigmentosa. Arch. Ophthalmol. 2009, 127, 784–790.
- Cideciyan, A.V.; Hood, D.C.; Huang, Y.; Banin, E.; Li, Z.Y.; Stone, E.M.; Milam, A.H.; Jacobson, S.G. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc. Natl. Acad. Sci. USA 1998, 95, 7103–7108.
- Li, J.; Edwards, P.C.; Burghammer, M.; Villa, C.; Schertler, G.F. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004, 343, 1409–1438.
- Iannaccone, A.; Man, D.; Waseem, N.; Jennings, B.J.; Ganapathiraju, M.; Gallaher, K.; Reese, E.; Bhattacharya, S.S.; Klein-Seetharaman, J. Retinitis pigmentosa associated with rhodopsin mutations: Correlation between phenotypic variability and molecular effects. Vision Res. 2006, 46, 4556–4567.
- Tuan-Phat Huynh; Shivani N. Mann; Nawajes A. Mandal; Botanical Compounds: Effects on Major Eye Diseases. Evidence-Based Complementary and Alternative Medicine 2013, 2013, 549174, 10.1155/2013/549174.
- Constance Saw; Yue Guo; Yuqing Yang; Ximena Paredes-Gonzalez; Christina Ramirez; Douglas Pung; Ah-Ng Tony Kong; The berry constituents quercetin, kaempferol, and pterostilbene synergistically attenuate reactive oxygen species: Involvement of the Nrf2-ARE signaling pathway. Food and Chemical Toxicology 2014, 72, 303-311, 10.1016/j.fct.2014.07.038.
- Virginia Miraldi Utz; Wanda Pfeifer; Susannah Q. Longmuir; Richard Olson; Kai Wang; Arlene Drack; Presentation of TRPM1-Associated Congenital Stationary Night Blindness in Children. JAMA Ophthalmology 2018, 136, 389-398, 10.1001/jamaophthalmol.2018.0185.
- Mandeep S. Singh; Robert E. MacLaren; Stem Cell Treatment for Age-Related Macular Degeneration: the Challenges. Investigative Opthalmology & Visual Science 2018, 59, AMD78-AMD82, 10.1167/iovs.18-24426.
- Joseph T. Ortega; Tanu Parmar; Beata Jastrzebska; Flavonoids enhance rod opsin stability, folding, and self-association by directly binding to ligand-free opsin and modulating its conformation. Journal of Biological Chemistry 2019, 294, 8101-8122, 10.1074/jbc.ra119.007808.
- Joseph T. Ortega; Tanu Parmar; Marcin Golczak; Beata Jastrzebska; Protective Effects of Flavonoids in Acute Models of Light-Induced Retinal Degeneration. Molecular Pharmacology 2020, 99, 60-77, 10.1124/molpharm.120.000072.
- Joseph T. Ortega; Beata Jastrzebska; The Retinoid and Non-Retinoid Ligands of the Rod Visual G Protein-Coupled Receptor. International Journal of Molecular Sciences 2019, 20, 6218, 10.3390/ijms20246218.
- Xiaoguang Cao; Melissa Liu; Jingsheng Tuo; Defen Shen; Chi-Chao Chan; The effects of quercetin in cultured human RPE cells under oxidative stress and in Ccl2/Cx3cr1 double deficient mice. Experimental Eye Research 2010, 91, 15-25, 10.1016/j.exer.2010.03.016.
- Minsup Lee; Seohyeon Yun; Hyesook Lee; Jaewook Yang; Quercetin Mitigates Inflammatory Responses Induced by Vascular Endothelial Growth Factor in Mouse Retinal Photoreceptor Cells through Suppression of Nuclear Factor Kappa B. International Journal of Molecular Sciences 2017, 18, 2497, 10.3390/ijms18112497.
- Sisi Weng; Lei Mao; Yuanyuan Gong; Tao Sun; Qing Gu; Role of quercetin in protecting ARPE-19 cells against H2O2-induced injury via nuclear factor erythroid 2 like 2 pathway activation and endoplasmic reticulum stress inhibition. Molecular Medicine Reports 2017, 16, 3461-3468, 10.3892/mmr.2017.6964.
- Daniel Kook; Armin H. Wolf; Alice L. Yu; Aljoscha S. Neubauer; Siegfried G. Priglinger; Anselm Kampik; Ulrich C. Welge-Lu¨ssen; The Protective Effect of Quercetin against Oxidative Stress in the Human RPE In Vitro. Investigative Opthalmology & Visual Science 2008, 49, 1712-1720, 10.1167/iovs.07-0477.
- Jun Kim; Hong Lan Jin; Dae Sik Jang; Kwang Won Jeong; Se-Young Choung; Quercetin-3-O-α-l-arabinopyranoside protects against retinal cell death via blue light-induced damage in human RPE cells and Balb-c mice. Food & Function 2018, 9, 2171-2183, 10.1039/c7fo01958k.
- Abolfazl Barzegar; Antioxidant activity of polyphenolic myricetin in vitro cell- free and cell-based systems. Molecular biology research communications 1970, 5, 87-95, .
- Minjuan Bian; Yong Zhang; Xiaoye Du; Jing Xu; Jingang Cui; Jiangping Gu; Weiliang Zhu; Teng Zhang; Yu Chen; Apigenin-7-diglucuronide protects retinas against bright light-induced photoreceptor degeneration through the inhibition of retinal oxidative stress and inflammation. Brain Research 2017, 1663, 141-150, 10.1016/j.brainres.2017.03.019.
- Wen-Wen Chou; Yung-Song Wang; Ku-Chung Chen; Jing-Mei Wu; Chung-Ling Liang; Suh-Hang Hank Juo; Tannic acid suppresses ultraviolet B-induced inflammatory signaling and complement factor B on human retinal pigment epithelial cells. Cellular Immunology 2012, 273, 79-84, 10.1016/j.cellimm.2011.11.003.
- Maria Hytti; Dora Szabó; Niina Piippo; Eveliina Korhonen; Paavo Honkakoski; Kai Kaarniranta; Goran Petrovski; Anu Kauppinen; Two dietary polyphenols, fisetin and luteolin, reduce inflammation but augment DNA damage-induced toxicity in human RPE cells. The Journal of Nutritional Biochemistry 2017, 42, 37-42, 10.1016/j.jnutbio.2016.12.014.
- Pascal Escher; Daniel F. Schorderet; Sandra Cottet; Altered Expression of the Transcription Factor Mef2c during Retinal Degeneration inRpe65–/–Mice. Investigative Opthalmology & Visual Science 2011, 52, 5933-5940, 10.1167/iovs.10-6978.
- Anne Wolf; Alexander Aslanidis; Thomas Langmann; Retinal expression and localization of Mef2c support its important role in photoreceptor gene expression. Biochemical and Biophysical Research Communications 2017, 483, 346-351, 10.1016/j.bbrc.2016.12.141.
- Daniela Calzia; Paolo Degan; Federico Caicci; Maurizio Bruschi; Lucia Manni; Luca A. Ramenghi; Giovanni Candiano; Carlo Enrico Traverso; Isabella Panfoli; Modulation of the rod outer segment aerobic metabolism diminishes the production of radicals due to light absorption. Free Radical Biology and Medicine 2018, 117, 110-118, 10.1016/j.freeradbiomed.2018.01.029.
- Lucio G. Costa; Jacqueline M. Garrick; Pamela J. Roquè; Claudia Pellacani; Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxidative Medicine and Cellular Longevity 2016, 2016, 2986796, 10.1155/2016/2986796.
- Joe G Hollyfield; Vera L Bonilha; Mary E Rayborn; Xiaoping Yang; Karen G Shadrach; Liang Lu; Rafael L Ufret; Robert G Salomon; Victor L Perez; Oxidative damage–induced inflammation initiates age-related macular degeneration. Nature Medicine 2008, 14, 194-198, 10.1038/nm1709.
- Moritz Veltmann; Margrit Hollborn; Andreas Reichenbach; Peter Wiedemann; Leon Kohen; Andreas Bringmann; Osmotic Induction of Angiogenic Growth Factor Expression in Human Retinal Pigment Epithelial Cells. PLOS ONE 2016, 11, e0147312-e0147312, 10.1371/journal.pone.0147312.
- Sun-Myung Yoon; Bom-Lee Lee; Yuan-Ri Guo; Se-Young Choung; Preventive effect of Vaccinium uliginosum L. extract and its fractions on age-related macular degeneration and its action mechanisms. Archives of Pharmacal Research 2016, 39, 21-32, 10.1007/s12272-015-0683-7.
- Yong Wang; Hye Jin Kim; Janet R. Sparrow; Quercetin and cyanidin-3-glucoside protect against photooxidation and photodegradation of A2E in retinal pigment epithelial cells. Experimental Eye Research 2017, 160, 45-55, 10.1016/j.exer.2017.04.010.
- Zhao Zhao; Tao Sun; Yun Jiang; Lijiang Wu; Xiangzhong Cai; Xiaodong Sun; Xiangjun Sun; Photooxidative damage in retinal pigment epithelial cells via GRP78 and the protective role of grape skin polyphenols. Food and Chemical Toxicology 2014, 74, 216-224, 10.1016/j.fct.2014.10.001.
- Ludmila F.M.F. Cardozo; Liliana M. Pedruzzi; Peter Stenvinkel; Milena Barcza Stockler-Pinto; Julio Daleprane; Maurilo Leite; Denise Mafra; Nutritional strategies to modulate inflammation and oxidative stress pathways via activation of the master antioxidant switch Nrf2. Biochimie 2013, 95, 1525-1533, 10.1016/j.biochi.2013.04.012.
- Amita Mishra; Amit Kumar Sharma; Shashank Kumar; Ajit K. Saxena; Abhay K. Pandey; Bauhinia variegataLeaf Extracts Exhibit Considerable Antibacterial, Antioxidant, and Anticancer Activities. BioMed Research International 2013, 2013, 915436, 10.1155/2013/915436.
- S Kumar; U K Sharma; A K Sharma; Abhay K Pandey; Protective efficacy of Solanum xanthocarpum root extracts against free radical damage: phytochemical analysis and antioxidant effect.. Cellular and molecular biology 2012, 58, 174-181, .
- Swapna Upadhyay; Madhulika Dixit; Role of Polyphenols and Other Phytochemicals on Molecular Signaling. Oxidative Medicine and Cellular Longevity 2015, 2015, 504253, 10.1155/2015/504253.
- Dyonne T Hartong; Eliot L Berson; Thaddeus P Dryja; Retinitis pigmentosa. The Lancet 2006, 368, 1795-1809, 10.1016/s0140-6736(06)69740-7.
- Hilda Petrs-Silva; Rafael Linden; Advances in gene therapy technologies to treat retinitis pigmentosa. Clinical Ophthalmology 2013, 8, 127-136, 10.2147/OPTH.S38041.
- Viviana Guadagni; Elena Novelli; Ilaria Piano; Maria Claudia Gargini; Enrica Strettoi; Pharmacological approaches to retinitis pigmentosa: A laboratory perspective. Progress in Retinal and Eye Research 2015, 48, 62-81, 10.1016/j.preteyeres.2015.06.005.
- Virginie Bernier; Daniel G Bichet; Michel Bouvier; Pharmacological chaperone action on G-protein-coupled receptors. Current Opinion in Pharmacology 2004, 4, 528-533, 10.1016/j.coph.2004.08.001.
- Gabriele D'Andrea; Quercetin: A flavonol with multifaceted therapeutic applications?. Fitoterapia 2015, 106, 256-271, 10.1016/j.fitote.2015.09.018.
- Tadao Maeda; Zhiqian Dong; Hui Jin; Osamu Sawada; Songqi Gao; Deepank Utkhede; Wendy Monk; Grazyna Palczewska; Krzysztof Palczewski; QLT091001, a 9-cis-Retinal Analog, Is Well-Tolerated by Retinas of Mice with Impaired Visual Cycles. Investigative Opthalmology & Visual Science 2013, 54, 455-466, 10.1167/iovs.12-11152.
- Tadao Maeda; Akiko Maeda; Gemma Casadesus; Krzysztof Palczewski; Philippe Margaron; Evaluation of 9-cis-Retinyl Acetate Therapy inRpe65−/−Mice. Investigative Opthalmology & Visual Science 2009, 50, 4368-4378, 10.1167/iovs.09-3700.
- J. Preston Van Hooser; Yan Liang; Tadao Maeda; Vladimir Kuksa; Geeng-Fu Jang; Yu-Guang He; Fred Rieke; Henry K. W. Fong; Peter Detwiler; Krzysztof Palczewski; et al. Recovery of Visual Functions in a Mouse Model of Leber Congenital Amaurosis. Journal of Biological Chemistry 2002, 277, 19173-19182, 10.1074/jbc.m112384200.