Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Phytochemicals in plant-based diets are thought to contribute substantially to lung cancer prevention and may be efficacious for targeting lung cancer stem cells.
- lung carcinogenesis
- phytochemicals
- cancer stem cell
- epigallocatechin-3-gallate
- sulforaphane
1. Introduction
Lung cancer is a malignant disease comprising two major histological categories known as non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC). In NSCLC, the major subtypes include adenocarcinoma (ADC), squamous cell carcinoma (SCC), and large cell carcinoma (LCC). A great proportion of cancer mortality today is still accounted for by lung cancer-related deaths (mainly NSCLC), representing 18.4% of all cancer deaths [1]. If proper preventive measures are not taken, about 1 in 5 people in countries with poor access to healthcare will die due to cancer by 2025 [2]. Some of the promoted cancer preventive measures include cessation of tobacco product and alcohol usage, pollution reduction, sufficient physical activity, and a healthy diet [3]. Among the proposed preventive solutions, eating a healthy diet that is mainly plant-based should be of primary concern, especially in developed countries, considering its huge contribution to overall health [4]. However, poor commitment due to various reasons such as high cost, poor taste, and low obtainability often renders this less achievable.
We generally acquire our nutrients from food in the form of macronutrients (i.e., carbohydrates, proteins, and fats) and micronutrients (i.e., vitamins and minerals). A plant-based diet that mainly consists of vegetables and fruits is considered good for well-being, mainly because it is enriched with vitamins, minerals, and dietary fibers. However, the contribution of micronutrients in vegetables and fruits to lowering the risk of lung cancer is controversial due to some inconsistent associations between the two variables [5,6]. Dietary fiber mainly helps in controlling other noncommunicable diseases such as cardiovascular disease, diabetes, and obesity [7]. Phytochemical compounds—the additional phytonutrients in vegetables and fruits—were found to have preventive and therapeutic potential for lung cancer as evidently shown by numerous in vitro and in vivo studies [8]. This suggests that phytochemical compounds may play a role in providing a plant-based diet’s added health benefits.
Cancer stem cell (CSC) theory postulates that there exists a tumor subpopulation for tumor maintenance, such as normal stem cells maintaining tissue steady state. CSC is the tumorigenic subpopulation capable of generating both tumorigenic and less tumorigenic offspring in the tumor bulk and is deemed responsible for the initiation and propagation of malignant diseases, including lung cancer [9]. This subpopulation is the reason why lung cancer gains resistance towards therapy over time and disease recurs in patients after initial remission [10]. Therefore, it is imperative to develop new therapies to target this subpopulation in order to achieve complete remission.
2. A Close Connection between Normal Lung Tissue Repair Mechanism and Carcinogenesis
The close resemblance of normal stem cells and CSCs is increasingly being recognized [11]. Both compartments are located at the apex of the hierarchy and possess high potency to proliferate and differentiate, thus making them capable of symmetric and asymmetric divisions. Both compartments also make use of respective stem cell niches for expansion cues and utilize similar signaling pathways in a tissue-specific manner [12,13]. Recently, we highlighted that lung progenitor cells could be the cells of origin of CSCs for different histological types of lung carcinomas, both of which are triggered to proliferate extensively in response to chronic injury [9]. In brief, basal cells, pulmonary neuroendocrine cells (PNECs), and alveolar epithelial cells II (AECs) are considered to be the cell origins of CSCs for SCC, SCLC, and ADC, respectively. It is not completely clear how these cells could evade a carcinogenic transformation and in what conditions these cells eventually become transformed. Nonetheless, these cells fulfill several criteria that accurately define how CSCs behave. In the sections below, the lungs and their resident epithelial cells, and how carcinogenesis can be triggered in those cells are briefly described, followed by a comprehensive description of how each resident cell proliferates and differentiates based upon the signaling pathways and chemical gradients they use during homeostasis maintenance and injury repair. This is important to recognize tissue- and cell-specific behavior that can be directly reflected towards the understandings of specific lung carcinomas.
2.1. Lung Tissue, Progenitor Cell, Cancer Stem Cell, and Injury-Triggered Carcinogenesis
The lung tissue consists of respiratory epithelia that line the trachea, bronchi, bronchioles, and alveoli for gas conduction and exchange (see Figure 1 for schematic representation). Major resident epithelial cells, such as goblet cells, ciliated cells, basal cells, and innervated PNECs, are found in the pseudostratified columnar ciliated layer of the epithelia, whereas club cells are found in the bronchiolar region [9,14]. Variant club cells that are resistant to the cytotoxic effects of naphthalene reside in the respiratory bronchioles along with PNECs clustered as the neuroepithelial bodies (NEB) and in the bronchioalveolar duct junctions [9,14]. AEC I and AEC II cells are restricted to the alveolar space [9,15]. Because the lungs are constantly exposed to pathogens and irritants via respiration, the resident epithelial cells have to ensure clearance of contaminants for homeostasis maintenance and inflammation evasion.
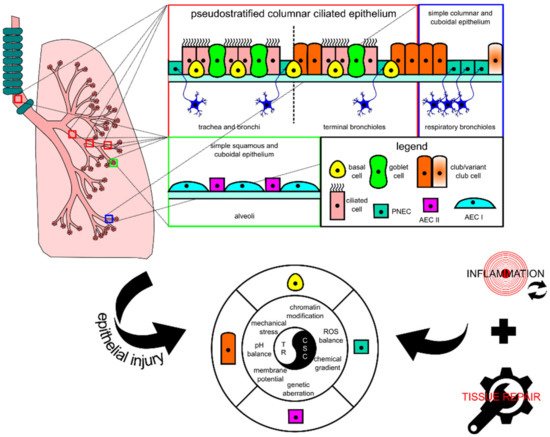
Figure 1. Schematic diagram showing the histological and cytological variations in different locations of the lung. In each tissue, there is at least one type of progenitor cell that is capable of proliferation, facilitating tissue repair in case of injury. In lung injury-triggered carcinogenesis, repeatedly induced inflammation accompanied by tissue repair could potentially lead to a chronic state that changes various extracellular and intracellular modulators and consequently encourages the emergence of lung cancer stem cells (CSCs) from progenitor cells. CSCs and tissue resolution (TR) are two sides of the same coin. AEC, alveolar epithelial cell; PNEC, pulmonary neuroendocrine cell; ROS, reactive oxygen species.
When the lungs suffer an injury, several lung resident cells such as basal cells, club cells, variant club cells, and AEC II cells could act as facultative stem cells to repair damage by repopulating lost cells [9]. PNECs can also take part in repopulating ciliated and club cells in naphthalene-induced injury [16]. These cells’ activity is lineage-committed, which means that their renewal function is induced at the respective location where they are residing. However, recent progress in the identification of progenitor cell candidates for lung injury repair points to the involvement of other cells, such as the lineage-negative epithelial progenitor (LNEP) cell, a basal cell progenitor termed the bronchial epithelial stem cell (BESC), and a subset of the AEC II cell population termed alveolar epithelial progenitor (AEP), in repairing distal lung epithelium [17,18,19]. These cells together with the well-studied facultative stem cells are key candidates for the emergence of CSCs because their proliferative activity is mostly injury-triggered and their activity coincides with other tumor-supporting events that occur after injury consisting of DNA damage and tissue repair-associated inflammation. Given the unique cellular turnover rate of lung epithelium and potent repair capacity during injury, these cells fulfill several premises to be defined as lung CSCs: (1) they are long-lived; (2) they are mostly quiescent; (3) they can survive mild or severe injury; (4) they are known to have the potency to differentiate and proliferate for both homeostasis and injury repair [9]. Based on the similarities in the location and protein markers, lung progenitors as key players for tissue repair have clear connections with CSCs of lung carcinomas, thus explaining the overlapping between tissue repair and carcinogenesis.
Continuous lung injury disrupts the normal physiology of the lungs by impairing the morphostatic field through systemic response such as chronic inflammation, which also damages DNA and proteins. Morphostatic components may include epigenetic sets of pre-programmed modules that determine cell fate decisions and tissue organization, including chromatin modifications, bioelectric events, mechanical stress, and chemical gradients [20]. Chronic inflammatory microenvironment may eventually result in wounds that do not re-pattern properly during wound repair, which is assumed to non-autonomously encourage CSC development and support tumor initiation [21]. Intracellular damage such as in DNA can be induced by genotoxic agents or through excess reactive oxygen species (ROS) generation also produced by inflammatory cells during injury-associated lung inflammation [22]. Prolonged DNA damage may lead the progenitor cells to accumulate genetic aberrations over time which could lead to tumorigenesis, and together with the epigenetic components, they may shape how DNA aberrations are recurring in a tissue-specific manner to maintain stemness, which also non-autonomously enhances tumor progression [21]. Thus, the emergence of CSCs is achieved through a series of processes that arrest tissue repair resolution and disrupt the morphostatic field, which enables the continuous proliferation of unregulated progenitor cells.
2.2. Normal Physiological Responses for Homeostasis and Injury Repair in Lung Tissue
To further understand how CSCs hijack the normal stem cell machinery for proliferation and differentiation, below we comprehensively describe the processes of homeostasis and how injury repair activities differ across different progenitor cells within the lung epithelia. The focus of the description is on morphostatic signaling pathways that regulate repair processes to maintain homeostasis, and their potential to control CSC activity.
At steady state, the lung tissue is actively maintained by the Hedgehog (Hh) pathway to remain in a quiescent state. During acute lung tissue injury, Sonic Hedgehog (SHH) protein is downregulated in SHH-expressing cells including club, ciliated, and AEC II cells, which consequently induces proliferation of adjacent GLI1+ SHH-responsive mesenchymal cells through a self-sufficient Platelet-Derived Growth Factor, B polypeptide (PDGFB)-dependent mechanism [23]. The absence of SHH paracrine signaling from lung epithelia removes the restriction to feed proliferative signals, i.e., FGF10 for lung epithelium regeneration [24].
2.2.1. Basal Cell
The basal cell is the most essential facultative stem cell marked by sex-determining region Y-box transcription factor 2 (SOX2), tumor protein 63 (TP63), and keratin 5 (KRT5) expressions. It is able to differentiate into ciliated, club, AEC I, and AEC II cells for homeostasis maintenance and injury repair [9]. A study in mice that combined long-term lineage tracing, biophysical modeling, and single-cell molecular analyses revealed that there were two pools of basal cells, comprising stem cell and committed progenitor pools [25]. SOX2 is required for basal cell specification from naïve epithelial cells as evident by transcriptional targeting of SOX2 on TP63 basal cell marker expression [26]. Besides TP63, the transcriptional coactivator YAP1 functions to maintain basal cell stemness by directly interacting with TP63 and regulating common target genes [27]. SOX2 is also used to maintain the basal cell’s identity and promote its self-renewal through Fibroblast Growth Factor Receptor 2 (FGFR2) signaling involving the Phosphoinositide 3-Kinase (PI3K)/AKT pathway [18,28].
During homeostasis maintenance, the basal cell is stochastically fated to differentiate into luminal lineages, and basal cell loss is compensated through duplication of neighboring basal cells [29]. The basal cell accomplishes club cell differentiation by generating the NOTCH3+ parabasal cell, which activates NOTCH1 and NOTCH2 by jagged 1 (JAG1) and JAG2 ligands [30]. Activation of Janus Kinase/Signal Transducer and Activator Transcription 3 (JAK/STAT3) signaling by interleukin 6 (IL6) from surrounding mesenchyme inhibits Notch signaling and directs the basal cell towards ciliated cell differentiation [31]. BMP ligand secretions from surrounding mesenchyme also reduce basal cell proliferation as shown by higher BMP antagonist Follistatin (FST) expression during regeneration [32].
2.2.2. Club Cell
The club cell is a prominent facultative stem cell within the bronchioles [33]. This cell is marked with SCGB1A1 marker expression and generated by basal cells and variant club cells during homeostasis and injury repair [34,35]. Similar to a basal cell, the club cell bears a proximal identity of SOX2 expression along with ciliated and goblet cells [36]. A club cell can also be regenerated by a variant club cell (SCGB1A1+cytochrome P450 2F2 (CYP2F2)+) through WNT7B-triggered paracrine stimulation of mesenchymal FGF10 resulting in simultaneous activation of Wnt/β-catenin and Notch pathways and transient Epithelial-to-Mesenchymal Transition (EMT) reprogramming in the epithelia, which in turn initiates repair [37].
For homeostasis and repair, the club cells are able to differentiate into ciliated cells by acting as transit-amplifying progenitor cells in the trachea and long-term progenitor cells in the bronchioles [38]. Alternate NOTCH2-JAG1-mediated lateral inhibition in neighboring cells directs to ciliated cell differentiation [39]. A recent lineage tracing assay and in vitro differentiation assay of purified club cells demonstrated wider club cells’ functional capability in repairing injured lung epithelia outside of the bronchiolar region by dedifferentiation into basal cells and transdifferentiation into AEC II cells to repair respective regional damage [18,40]. Interestingly, tumor protein 53 (TP53) also affects club cell proliferation and differentiation by keeping epithelial quiescence at steady state [41]. Loss of expression of TP53 leads to poorer differentiation of club cell to ciliated cell but higher dedifferentiation potential, e.g., self-renewing or dedifferentiating to a more naïve cell fate such as TP63+KRT5- progenitor cells, which can normally be inhibited through contact inhibition imposed by a neighboring basal cell [41,42]. YAP1 overexpression phenocopies TP53 loss in a club cell [27]. This may suggest that Hippo/YAP1 signaling and TP53 are somehow interacting to achieve balance to maintain lung epithelial quiescence by controlling the tendency to proliferate and differentiate [43].
2.2.3. PNEC
The PNEC is marked by Achaete-Scute homolog 1 (ASCL1), calcitonin gene-related peptide 1 (CALCA), synaptophysin (SYP), chromogranin A (CHGA), and ubiquitin carboxy-terminal hydrolase isozyme L1 (UCHL1) [16]. While it is not previously known to be derived from any progenitor cell in the lung, a PNEC can be generated by a SHH-irresponsive Twist-related protein 2 (TWIST2)+ mesenchymal cell on the occasion when the lung epithelia suffer naphthalene or lipopolysaccharide-induced injury [44]. Apart from renewing themselves, PNECs can repopulate club and ciliated cells in dire situations [16]. UCHL1+ PNECs can serve to repopulate club cells when wiped out by naphthalene treatment through a NOTCH1-dependent Notch signaling induction [45]. Transdifferentiation of PNEC to the ciliated cell uses the histone-lysine N-methyltransferase EZH2-IL6-STAT3 pathway [46]. A recent study discovered that a small number (2–4 cells) of differentiated PNECs in a cluster of NEB (a group of 20–30 PNECs) possess stem cell function, thus representing dedicated stem cells that are capable of self-renewal and differentiation under the strict guidance of genes such as transformation-related protein 53 (Trp53) and retinoblastoma (Rb) (quiescent signals) to ensure injury-dependent proliferation and control of subsequent sensitivity to dispersal and transdifferentiation signals [47].
2.2.4. AEC II
AEC II is a prominent facultative stem cell in the peripheral lung epithelia. This cell is marked with the expression of pulmonary Surfactant-associated protein C (SFTPC) and mainly only self-renews and differentiates into Homeodomain-only protein (HOPX)+Podoplanin (PDPN)+Aquaporin 5 (AQP5)+ AEC I for homeostasis maintenance [15]. AEC II is known to have restricted regenerative capacity that only generates residents within the alveolar compartment for homeostasis and mild injury repair. To maintain the integrity of the alveolar compartment, neo-basal/basal-like cells could be generated by several precursors or intermediate progenitor cells to repair severe injury, including LNEPs, BESCs, SOX2+ progenitor cells, and intrapulmonary bronchial progenitor cells [17,18,48,49]. In contrast to the basal cell in the proximal region, neo-basal/basal-like cells generated by LNEPs are maintained by hypoxia-inducible factor 1-alpha (HIF1A)-mediated Notch activity and differentiate into AEC II upon Wnt/β-catenin-driven Notch blockade [17,50]. On the other hand, BESCs are also capable of differentiating to neo-basal/basal-like cells that further differentiate into AEC II or club cells in response to bleomycin-induced severe lung injury [18].
FGF10-FGFR2B signaling strength controls whether the BESC-generated neo-basal/basal-like cells are expanded or transdifferentiated to AEC II; a sufficiently high level of FGF10 biases towards AEC II differentiation, whereas subsequent FGF10 supply termination leads to AEC I differentiation [18]. The sufficiency of FGF10 feed is controlled by epithelial WNT7B feedback to airway smooth muscle cells (ASMCs), which is negatively controlled by the Integrin-Linked Kinase (ILK)-Hippo pathway for quiescence maintenance [51]. Apart from being responsive to FGF10 signaling, AEC II also proliferates in response to EGFR and GTPase KRAS signaling [52]. A single Platelet-Derived Growth Factor Receptor Alpha (PDGFRA)+ fibroblast can serve as the stem cell niche for an AEC II by supplying Wnt ligands that allow the cell to proliferate extensively with appropriate signals and maintain its stem cell identity, which would otherwise be differentiated to AEC I upon exiting the niche [53]. This Wnt active population (AXIN2+Transmembrane 4 L6 family member 1 (TM4SF1)+) is a distinct smaller AEC II cell population termed AEPs [19]. Under acute injury response, AEPs become Wnt autocrine-proficient, thus enabling them to regenerate lost AEC I and AEC II [53]. Like how BMP signaling influences the basal cell, the stable expression of phosphorylated mothers against decapentaplegic homolog 1/5/8 (pSMAD 1/5/8) of BMP signaling in AEC I and AEC II maintains cellular quiescence, whereas expression of antagonist FST promotes AEC II proliferation [54].
The signaling pathways regulating self-renewal and overall interactions of lung epithelial residents for proliferation and differentiation are simplistically summarized in Figure 2.
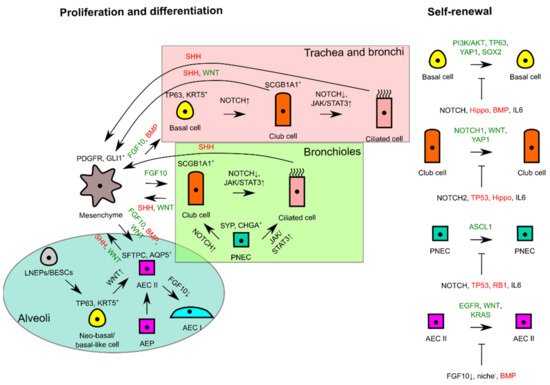
Figure 2. Schematic diagram illustrating self-renewal and the interactions of lung epithelial residents with mesenchyme to trigger proliferation or to maintain tissue quiescence. In the different proximodistal locations of lung epithelia, signaling pathways are used differently for differentiation, whereas similar signals are used for proliferation and lung tissue quiescence in progenitor cells of respective lung tissue locations. FGF10 or WNT from mesenchyme consequently triggers self-renewal of progenitor cells if differentiating and tissue quiescence signals (--|sign) are missing during tissue repair or homeostasis. Green signals favor proliferation, whereas red signals favor tissue quiescence. AEC, alveolar epithelial cell; AEP, alveolar epithelial progenitor; BESC, bronchial epithelial stem cell; LNEP, lineage-negative epithelial progenitor; PNEC, pulmonary neuroendocrine cell.
(References would be added automatically after the entry is online)
This entry is adapted from the peer-reviewed paper 10.3390/ijms22115697
This entry is offline, you can click here to edit this entry!