Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
|
Neurosciences
Conditions such as Alzheimer’s (AD) and Parkinson’s diseases (PD) are less prevalent in cancer survivors and, overall, cancer is less prevalent in subjects with these neurodegenerative disorders. In addition to epidemiologic data, there is also evidence of a complex biological interconnection, with genes, proteins, and pathways often showing opposite dysregulation in cancer and neurodegenerative diseases.
- cancer
- neurodegeneration
- inverse comorbidity
1. Cancer and Neurodegenerative Diseases
The World Health Organization (WHO) highlights cancer as one of the most common causes of death, which accounted for almost 10 million deaths worldwide in 2020 [1]. Overall, estimates indicate that one in five persons will get cancer in their lifetime before 75 years of age and one in ten will die from the disease.
The incidence and prevalence of neurodegenerative diseases are also high. Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by brain β-amyloid plaques and neurofibrillary tangles formed by phosphorylated tau (P-Tau) protein deposits. AD is the most frequent neurodegenerative disease and affects 24 million people worldwide. The second most frequent neurodegenerative disease is Parkinson’s disease (PD), characterized by Lewy bodies and neurites formed by alpha-synuclein deposits. PD has a prevalence of 1% in people older than age 60, and of 3% in people aged 80 years or older [2]. There is also increasing interest, in the literature, on multiple sclerosis (MS). Despite being an inflammatory demyelinating disease, MS can also be viewed as a neurodegenerative disease because of the cascade of events triggered by neuroinflammation and leading to neurodegeneration [3][4][5]. The key elements that induce neurodegeneration include activation of microglia, chronic oxidative damage, and altered mitochondrial function in axons, leading to chronic cellular stress and imbalance of ion homeostasis, resulting in axonal and neuronal death [6]. The total prevalence of MS in Europe is 83 per 100,000, which is lower than the prevalence of AD and PD, but still associated with substantial social and economic costs [7]. Amyotrophic lateral sclerosis (ALS), which is characterized by the progressive loss of motor neurons in the brain and spinal cord, is also rare, with a prevalence of 2–3 per 100,000 [8].
2. The Connection between Cancer and Neurodegenerative Diseases
The incidence of many common cancers and neurodegenerative diseases including AD and PD increases with age [9][10]. MS also typically occurs in adults, although its prevalence peaks for subjects between 35 and 64 years of age and does not further increase thereafter [7]. Similarly, the risk for ALS peaks at 50–75 years of age [8]. It is essential to focus attention on the development of alternative treatments that address age-related diseases, also in consideration of the increase in age in the population.
Despite the common age-related trends in the incidence of cancer and neurodegenerative disorders, a meta-analysis of observational studies including 577,013 participants concluded that there was a significantly lower co-occurrence of cancer in patients with neurodegenerative disorders. In particular, patients with AD had a markedly reduced co-occurrence of cancer in general, but no data were available for specific cancers [11]. The Framingham heart study, a longitudinal community based cohort study, in which 221 cases were evaluated for a 10-year follow-up, also indicated a lower risk of AD for cancer survivors: the risk of AD was lower in survivors of smoking-related cancers; this model for cancer is similar to that seen in PD and suggests an inverse association between cancer and neurodegeneration; the effect was stronger for lung cancer and preserved for participants who survived at least to 80 years of age [12]. Furthermore, in evaluating the interconnection between AD and lung cancer, it is important to consider that cigarette smoking appears to play a neuroprotective influence for both AD and PD [13], while it represents a known risk factor for cancer of the lung.
A recent systematic review and meta-analysis confirmed a weak but significant decrease in AD risk comparing older adults with vs. without a previous cancer diagnosis, but it could not rule out a role of survival bias [14]. Epidemiological studies indicate that AD patients have a lower risk of developing lung cancer and a higher risk of developing glioblastoma: transcriptomic meta-analyses reveal a significant number of genes with reverse expression patterns in AD and lung cancer, compared to AD and glioblastoma [15]. Meta-analytical data indicate that patients with PD and MS have a reduced co-occurrence of lung and prostate cancers, and patients with PD also have reduced co-occurrence of colorectal cancer. These associations are not consistent across all cancer types, with an increased cooccurrence of melanoma in patients with PD and of brain cancers in patients with MS [11]. On the other hand, a recent population-based case-control study reported negative point estimates of the odds of developing PD in survivors of most cancers, with the notable exception of skin and female breast cancer but with wide confidence intervals overlapping with zero [16]. A recent meta-analysis has shown that patients with MS have a lower risk of contracting tumors than the general population [17], with an inverse comorbidity between MS and cancer. The authors also stressed that the identification of inverse comorbidity and its underlying mechanisms could provide important new insights into the understanding of MS [18]. No significant association has been found between ALS and overall cancer occurrence [11].
These epidemiologic associations are complex and challenged by confounders and exceptions [19]. Cancer treatment may also modify the relationship; some studies suggest that patients who received chemotherapy may have lower white matter organization and connectivity when compared with healthy controls [20]. Other studies suggest that chemotherapy correlates with a lower risk of AD [21]. Inverse comorbidity does not involve all types of neurodegenerative disorders and all types of cancers. Thus, the concept of inverse comorbidity, as it is discussed in this review, should always be considered in its imperfect nature rather than as a fixed rule. Nevertheless, the epidemiological data suggest a pattern of lower-than-expected combined probability of cancer and neurodegenerative diseases, which has been defined as “inverse comorbidity” [22]. The study of the mechanisms behind this pattern of inverse comorbidity could influence therapeutic interventions and provide strategies that prevent or delay both cancer and neurodegenerative diseases [19][23][24][25].
There are multiple factors that play a central role both in cancers and neurodegeneration through the same metabolic pathways that are inversely regulated and altered, as outlined in a comprehensive review published in recent years [19]. One key feature highlighted by that review was the occurrence of two patterns of association between cancer and neurodegeneration, which were summarized as “proliferation” and “apoptosis”, respectively. This implied that neurodegenerative diseases and cancers may be viewed as two sets of diseases with “too little” and “too much” apoptosis, respectively. The review also highlighted multiple factors that may underlie this difference: oxidative stress, DNA damage, inflammation, genomic instability and epigenetic alterations, mitochondrial and telomere dysfunction, metabolic dysregulation, depletion of stem cells, aberrant activation of the cell cycle, and cellular interconnections [19]. Many of these factors overlap with those that have been proposed as the pillars of aging: macromolecular damage, proteostasis, inflammation, epigenetics, metabolism, stem cells and regeneration, and adaptation to stress [26].
Another potential example of a mechanism relevant to both cancer and neurodegeneration is the non-classical, non-enzymatic binding of acetylcholinesterase (AChE) acting at an allosteric site on the nicotinic alpha-7 receptor. AChE is expressed not only in the brain but also in epithelial, endothelial, immune, and cancer cells. This form of inter-cellular communication may represent a system for triggering the entry of calcium into a wide range of cells to promote their growth. Furthermore, the AChE peptide might play a fundamental role in cell migration; therefore, through this pathway (common to neurodegeneration and carcinogenesis), there might be an interconnection between the nervous, endocrine, and immune control systems [27].
Although neurodegeneration is typical of old age, neurodevelopmental disorders might share at least some mechanisms with neurodegenerative diseases, with cognitive delay representing the counterpart of dementia. It has been estimated that 40–100% of brain tumor survivors have neurocognitive problems [28], not necessarily connected with the direct brain damage associated with the tumor and/or its treatment, and a recent review has highlighted the bidirectional correlation between cancer and neurodevelopmental disorders in pediatric age [29]. Although pediatric studies conducted on this topic are still very few, a role in the neurodevelopment deficit (especially in a fundamental period for cerebral maturation and neuronal plasticity) is played by anticancer therapies, due to their neurotoxicity [30]. Over the years, various chemotherapeutic agents used in clinical practice for the treatment of brain tumors have shown severe effects on cognitive functions, and recent studies on nanotechnology have shown that nanomaterials that could be exploited for the treatment of brain cancer are able to induce neurotoxic effects and neurodegeneration [31]. However, another important role is definitely played by inflammatory processes related to the tumor and to the considerable increase of reactive oxygen species (ROS); thus, cytokine-mediated inflammatory mechanisms might act as a trigger that initiates a cascade of events responsible for neurotoxicity [32]. In support of this, a study on children with acute lymphatic leukemia showed damage to brain structures both before and after chemotherapy, with an increase in Tau protein (suggestive of axonal damage) and in glial fibrillary acidic protein (GFAP) in patients with alterations of the apolipoprotein E gene (associated with attention deficit) and an increase in leukoencephalopathy, with impairment of the white matter [32][33].
3. The Biological Bases of the Inverse Comorbidity between Cancer and Neurodegeneration
Inverse comorbidity between cancer and neurodegeneration may be influenced by environmental, pharmacological, and dietary factors. Genetic factors can additionally contribute to the inverse comorbidity between complex diseases [22][34][35][36]. At the base of the bidirectional interactions between cancer and neurodegenerative diseases there may be complex mechanisms that include genes, proteins, and mitochondrial function, the study of which could provide important therapeutic novelties for both diseases (Figure 1).
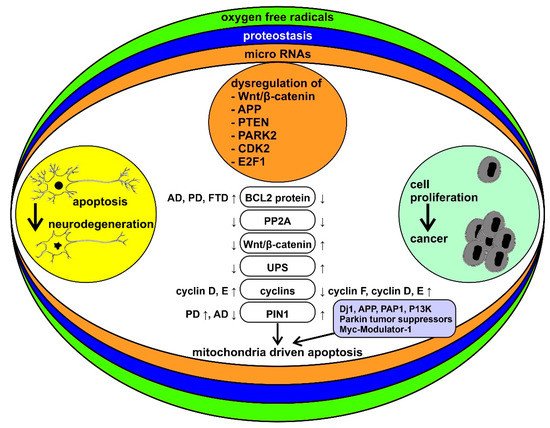
Figure 1. Schematic representation of the bidirectional interactions between cancer and neurodegenerative diseases and their modulatory processes. Neurodegenerative diseases and cancer are framed as two sets of disorders with “too much” or “too little” apoptosis, respectively. Reactive oxygen species (ROS), alterations in proteostasis, and microRNA (miRNAs) are among the key factors that may underlie the differences between the two sets of disorders. Some of the specific molecular mechanisms thought to be involved in this pattern of inverse comorbidity are highlighted in the middle column (see the text of the paper for abbreviations). ↓ = downregulated, ↑ = upregulated.
Mutations in PRKN (PARK2, Parkin), PARK7 (DJ-1), and PINK1 genes, which are among the genes contributing to familial cases of PD, lead to the mutation of both tumor suppressor genes, PTEN and TP53 [25]. On the other hand, PRKN and PARK5 have antiproliferative properties and are often inactivated in tumors [37]; PINK1 can also have antiproliferative functions [38]. Another mechanism potentially contributing to the inverse comorbidity is related to the brain expression of PARP1. The PARP1 protein is underexpressed in the brains of subjects with PD [39], while it is overexpressed in glioblastoma multiforme [39]. Interestingly, the PARP1 protein is also overexpressed in prostate cancer [40].
The genetic variants shared between AD and cancer are less known. Recently, a genome-wide association study (GWAS) has highlighted genes potentially involved in AD and in five different cancers (colon, breast, prostate, ovary, lung), with some shared variants modulating disease risk in a concordant way and others exerting effects in opposite directions [41]. On the other hand, the transcription factor P53 (a tumor suppressor) has multiple functions common to cancer and neurodegenerative disorders such as HD, PD, and AD [25][42], and it is crucial for cell growth control and apoptosis. Its expression is upregulated in AD, PD, and HD but downregulated in the vast majority of tumors [19][23][25].
Aggregation of superoxide dismutase (SOD1) causes cell death in ALS; however, SOD1 also has a role in breast cancer and the ability to increase estrogen reactive gene expression [19]. The reduced activity of SOD1 and glutathione reductase (GR) induces an increase in the production of reactive oxygen species (ROS), which leads to a conformational change of P53. The modification of P53, in turn, induces the production of ROS, thus activating oncogenic functions, such as tumor cell invasion and metastases. P53 also induces the upregulation of apoptotic proteins, such as X associated with BCL2 (BAX) and caspase 3 (observed in PD) [43]. This cascade of events, depicted also in Figure 2, seems to be an example of unclear or absent inverse comorbidity between neurodegeneration and cancer, perhaps based on the physiological and ubiquitous ROS signaling.
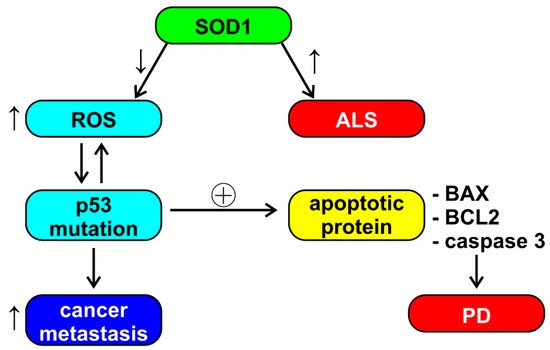
Figure 2. Schematic representation of the possible role of superoxide dismutase (SOD1) in amyothrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and cancer metastasis. See the text of the paper for abbreviations. ↓ = decrease, ↑ = increase, ⊕ = stimulation.
Interestingly, drugs used in the treatment of symptoms of neurodegenerative diseases, such as thioridazine, have been shown to have anticancer effects, while anticancer drugs, such as cyclin-dependent kinase inhibitors and mithramycin, are neuroprotective; these data reinforce the existence of a link between cancer and central nervous system (CNS) diseases and indicate that future studies will need to focus on specific molecular pathways [23].
4. The Role of Mitochondria
The BCL2 protein, involved in the mitochondrial outer membrane permeabilization, is overexpressed in CNS disorders, such as AD, PD, and frontotemporal dementia (FTD), whereas it undergoes downregulation in tumors [23]. On the other hand, cyclin D and cyclin E are overexpressed in both cancer and neurodegenerative diseases, while PP2A is downregulated in both diseases. Cyclin F is downregulated in cancer, and a mutation has been found in neurodegenerative diseases [43]. PIN1, a multifunctional gene that is hypothesized to function as a molecular timer, is overexpressed in a number of tumors and in PD but is underexpressed in AD [19][43]. Inhibition of PIN1 suppresses the growth of various tumor cells and is therefore considered as a promising therapeutic target in the oncological field [43]. PIN1, through its interactions with P53 and BCL2, can have a pro- or anti-apoptotic role depending on the cellular context. Its role in mitochondria-driven apoptosis could therefore provide a link between cancer and AD [43]. Other potentially common factors in cancer and neurodegenerative diseases include the PARK7 (DJ-1) and APP oncogenes (the first is involved in mitochondrial regulation, the second is a precursor of the β-amyloid protein), the PFDN5 (MM-1) and PRKN (Parkin) tumor suppressors (the first inhibits transcription and protein aggregation, involved in the onset of PD, cerebellar atrophy and HD, the latter plays a role in mitophagy and ubiquitination), and the PDAP1 (PAP1) and PINK1 modulators (the former is involved in splicing and the latter in mitophagy) [44].
Growing evidence suggests that age-related changes in bioenergetics at the mitochondrial level and the resulting metabolic compensation may be an important driver of both cancer and neurodegeneration and a potential target for prevention and therapy; dysfunction of mitochondria leads to disruption of DNA repair and the malfunction of metabolic pathways (such as the PI3K pathway), increasing the risk of cancer [45]. Alterations in mitochondrial DNA (mtDNA) decrease the efficiency of the respiratory chain with aging, and the prevalence of mtDNA deletions seems particularly high in neurodegenerative disorders such as PD and AD [46][47]. It has also been shown that mutations in mitochondrial DNA and alterations in mitochondrial energy metabolism can be correlated with the onset or progression of glioblastoma, following alteration of the pathways involved in apoptosis [48].
Recent data highlight an important role of the intestinal microbiome, intestinal permeability, and alterations in the functioning of mitochondria in the pathophysiology of MS: orexins, melatonin, and butyrate increase oxidative phosphorylation in mitochondria through the disinhibition of the pyruvate dehydrogenase complex, leading to an increase in acetyl-coenzyme A, a co-substrate necessary for the activation of the melatoninergic pathway of the mitochondria. Loss of mitochondrial melatonin coupled with an increase in N-acetylserotonin has implications for impaired mitochondrial function and appears to play a role in the pathophysiology of MS [49].
5. Other Factors
Telomere alterations have been recognized as a risk factor for both age-related carcinogenesis and neurodegeneration. In AD, telomere shortening has been implicated in oxidative stress and inflammation, with cognitive impairment, amyloid pathology, and hyperphosphorylation of the Tau protein. A decrease in telomere length was found in peripheral blood leukocytes in patients with AD, compared with age-matched controls, and was proposed as a potential biomarker for AD [50][51]. As introduced above, this should be considered in the framework of the imperfect inverse comorbidity between AD and cancer. Several studies have shown that telomeres shorten in the early stages of carcinogenesis and that tumor cells need to activate telomerases (which synthesize telomeres) to become immortal [52][53].
The Wnt family of secreted glycolipoprotein signaling pathway molecules takes part in the regulation of cell proliferation, polarity, and fate during the embryonic phase and in tissue homeostasis. Changes in the Wnt pathway are involved in congenital defects, cancer, and other diseases [54]. The Wnt/β-catenin signaling pathway appears to play a critical role in neural stem cell proliferation [55]. However, the dysregulation of this pathway has also been associated with cancer and neurodegenerative disorders, such as AD and PD, through an inverse correlation. In particular, the activation of Wnt signaling could be protective in neurodegenerative diseases but could promote the onset and progression of cancer [56][57]. Several molecular components of the signaling pathway have been proposed as innovative targets for cancer therapy, and very recently, some have also been evaluated as potential therapeutic targets for PD [57].
Protein homeostasis (or proteostasis) indicates the maintenance of the correct concentration of proteins of regular conformation and subcellular compartmentalization. The loss of protein homeostasis is another very important process in neurodegeneration. In particular, the family of heat shock proteins, chaperones, and the ubiquitin–proteasome system (UPS), which decrease with aging, result in aggregation of synuclein in Lewy bodies, of β-amyloid, and of Tau [58][59]. Additionally, neoplastic cells show a loss of protein homeostasis but often in the opposite direction: in cancer cells there is an overexpression of UPS and heat shock proteins [60][61]. Global hypomethylation and hypohydroxymethylation, alterations of histone proteins, and high expression of some non-coding RNAs were found in AD [62][63]. The same mechanisms are also implicated in carcinogenesis, and epigenetic therapy has already been suggested as a potential method to correct the expression levels of dysregulated genes in neurodegenerative disorders and tumors [19].
The miR-34 and miR-122 miRNAs have been used in the treatment of certain types of cancer and hepatitis, with promising results [64]. On the other hand, some miRNAs (miRNA-9, miRNA-34a, miRNA-125b, miRNA-146a, and miRNA-155) may be involved in the physiopathology of AD, and some act on the Wnt/β-catenin pathway [65], which is involved in both neurodegenerative diseases and tumors. A recent paper suggested a potential anti-β-amyloid protective effect of miRNA-15b and a biological link between miRNA-125b and neurotoxic pathways, hypothesizing that these miRNAs may play a role as biomarkers of AD physiopathology with therapeutic potential [66]. The amyloid precursor protein (APP) is connected to both AD and malignant growth. APP is concentrated in neuronal synapses and is the major component of amyloid plaques associated with AD. APP increases the proliferation and migration of epithelial cells (although the mechanism has not been fully defined) and is overexpressed in various tumors (oral cavity, esophagus, pancreatic, neuroendocrine, thyroid, and colorectal cancers) [67]. These results suggest the potential role of APP in cancer pathogenesis and reinforce our concept of imperfect inverse comorbidity between AD and cancer. Aberrant expression of miRNAs could also be involved in both neurodegeneration and tumor pathologies through the downregulation of PTEN, involved in PD. Many genes involved in both types of pathologies, including PARK2, CDK2, and E2F1, are potential targets of multiple miRNAs; however, further studies are needed to better understand their roles [67]. Taken together, these data suggest that miRNAs may be the basis for common therapeutic approaches to both cancer and neurodegenerative diseases [67][68].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13112612
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021.
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10.
- Trapp, B.D.; Nave, K.-A. Multiple Sclerosis: An Immune or Neurodegenerative Disorder? Annu. Rev. Neurosci. 2008, 31, 247–269.
- Giovannoni, G. The neurodegenerative prodrome in multiple sclerosis. Lancet Neurol. 2017, 16, 413–414.
- Chaudhuri, A. Multiple sclerosis is primarily a neurodegenerative disease. J. Neural Transm. 2013, 120, 1463–1466.
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193.
- Pugliatti, M.; Rosati, G.; Carton, H.; Riise, T.; Drulovic, J.; Vecsei, L.; Milanov, I. The epidemiology of multiple sclerosis in Europe. Eur. J. Neurol. 2006, 13, 700–722.
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098.
- Palmer, S.; Albergante, L.; Blackburn, C.C.; Newman, T.J. Thymic involution and rising disease incidence with age. Proc. Natl. Acad. Sci. USA 2018, 115, 1883–1888.
- de Pedro-Cuesta, J.; Rabano, A.; Martinez-Martin, P.; Ruiz-Tovar, M.; Alcalde-Cabero, E.; Almazan-Isla, J.; Avellanal, F.; Calero, M. Comparative Incidence of Conformational, Neurodegenerative Disorders. PLoS ONE 2015, 10, e0137342.
- Catalá-López, F.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Valderas, J.M.; Gómez-Beneyto, M.; Martinez, S.; Balanzá-Martínez, V.; Climent, J.; Valencia, A.; McGrath, J.; et al. Inverse and Direct Cancer Comorbidity in People with Central Nervous System Disorders: A Meta-Analysis of Cancer Incidence in 577,013 Participants of 50 Observational Studies. Psychother. Psychosom. 2014, 83, 89–105.
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse Association between Cancer and Alzheimer’s Disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442.
- Fratiglioni, L.; Wang, H.-X. Smoking and Parkinson’s and Alzheimer’s disease: Review of the epidemiological studies. Behav. Brain Res. 2000, 113, 117–120.
- Ospina-Romero, M.; Glymour, M.M.; Hayes-Larson, E.; Mayeda, E.R.; Graff, R.E.; Brenowitz, W.D.; Ackley, S.F.; Witte, J.S.; Kobayashi, L.C. Association between Alzheimer Disease and Cancer with Evaluation of Study Biases: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2020, 3, e2025515.
- Sánchez-Valle, J.; Tejero, H.; Ibáñez, K.; Portero, J.L.; Krallinger, M.; Al-Shahrour, F.; Tabares-Seisdedos, R.; Baudot, A.; Valencia, A. A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer’s Disease, Glioblastoma and Lung cancer. Sci. Rep. 2017, 7, 4474.
- Cui, X.; Liew, Z.; Hansen, J.; Lee, P.-C.; Arah, O.A.; Ritz, B. Cancers Preceding Parkinson’s Disease after Adjustment for Bias in a Danish Population-Based Case-Control Study. Neuroepidemiology 2019, 52, 136–143.
- Ghajarzadeh, M.; Mohammadi, A.; Sahraian, M.A. Risk of cancer in multiple sclerosis (MS): A systematic review and meta-analysis. Autoimmun. Rev. 2020, 19, 102650.
- Thormann, A.; Koch-Henriksen, N.; Laursen, B.; Sørensen, P.S.; Magyari, M. Inverse comorbidity in multiple sclerosis: Findings in a complete nationwide cohort. Mult. Scler. Relat. Disord. 2016, 10, 181–186.
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2018, 11, 77–89.
- Kesler, S.R.; Watson, C.L.; Blayney, D.W. Brain network alterations and vulnerability to simulated neurodegeneration in breast cancer. Neurobiol. Aging 2015, 36, 2429–2442.
- Frain, L.; Swanson, D.; Cho, K.; Gagnon, D.; Lu, K.P.; Betensky, R.A.; Driver, J. Association of Cancer and Alzheimer’s Disease Risk in a National Cohort of Veterans. Alzheimer’s Dement. 2017, 13, 1364–1370.
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013, 14, 293–304.
- Klus, P.; Cirillo, D.; Orfila, T.B.; Tartaglia, G.G. Neurodegeneration and Cancer: Where the Disorder Prevails. Sci. Rep. 2015, 5, 15390.
- Tallaksen, C.M.; Muller, U. Cancer and Neurodegeneration: Time to Move Beyond Janus? Neurology 2017, 88, 1106–1107.
- Rojas, N.G.; Cesarini, M.; Etcheverry, J.L.; Da Prat, G.A.; Arciuch, V.A.; Gatto, E.M. Neurodegenerative diseases and cancer: Sharing common mechanisms in complex interactions. J. Integr. Neurosci. 2020, 19, 187–199.
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713.
- Garcia-Ratés, S.; Greenfield, S. Cancer and neurodegeneration: Two sides, same coin? Oncotarget 2017, 8, 22307–22308.
- Duffner, P.K. Risk factors for cognitive decline in children treated for brain tumors. Eur. J. Paediatr. Neurol. 2010, 14, 106–115.
- Mogavero, M.P.; Bruni, O.; DelRosso, L.M.; Ferri, R. Neurodevelopmental Consequences of Pediatric Cancer and Its Treatment: The Role of Sleep. Brain Sci. 2020, 10, 411.
- Marusak, H.A.; Iadipaolo, A.S.; Harper, F.W.; Elrahal, F.; Taub, J.W.; Goldberg, E.; Rabinak, C.A. Neurodevelopmental consequences of pediatric cancer and its treatment: Applying an early adversity framework to understanding cognitive, behavioral, and emotional outcomes. Neuropsychol. Rev. 2018, 28, 123–175.
- Catalan-Figueroa, J.; Palma-Florez, S.; Álvarez, G.; Fritz, H.F.; O Jara, M.; O Morales, J. Nanomedicine and nanotoxicology: The pros and cons for neurodegeneration and brain cancer. Nanomedicine 2016, 11, 171–187.
- Cheung, Y.T.; Brinkman, T.M.; Mulrooney, D.A.; Mzayek, Y.; Liu, W.; Banerjee, P.; Panoskaltsis-Mortari, A.; Srivastava, D.; Pui, C.-H.; Robison, L.L.; et al. Impact of sleep, fatigue, and systemic inflammation on neurocognitive and behavioral outcomes in long-term survivors of childhood acute lymphoblastic leukemia. Cancer 2017, 123, 3410–3419.
- Cheung, Y.T.; Khan, R.B.; Liu, W.; Brinkman, T.M.; Edelmann, M.N.; Reddick, W.E.; Pei, D.; Panoskaltsis-Mortari, A.; Srivastava, D.; Cheng, C.; et al. Association of Cerebrospinal Fluid Biomarkers of Central Nervous System Injury with Neurocognitive and Brain Imaging Outcomes in Children Receiving Chemotherapy for Acute Lymphoblastic Leukemia. JAMA Oncol. 2018, 4, e180089.
- Tabarés-Seisdedos, R.; Dumont, N.; Baudot, A.; Valderas, J.M.; Climent, J.; Valencia, A.; Crespo-Facorro, B.; Vieta, E.; Gómez-Beneyto, M.; Martinez, S.; et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol. 2011, 12, 604–608.
- Devine, M.J.; Plun-Favreau, H.; Wood, N.W. Parkinson’s Disease and Cancer: Two Wars, One Front. Nat. Rev. Cancer 2011, 11, 812–823.
- West, A.B.; Dawson, V.L.; Dawson, T.M. To Die or Grow: Parkinson’s Disease and Cancer. Trends Neurosci. 2005, 28, 348–352.
- Inzelberg, R.; Samuels, Y.; Azizi, E.; Qutob, N.; Inzelberg, L.; Domany, E.; Schechtman, E.; Friedman, E. Parkinson disease (PARK) genes are somatically mutated in cutaneous melanoma. Neurol. Genet. 2016, 2, e70.
- Matsushima-Nishiu, M.; Unoki, M.; Ono, K.; Tsunoda, T.; Minaguchi, T.; Kuramoto, H.; Nishida, M.; Satoh, T.; Tanaka, T.; Nakamura, Y. Growth and gene expression profile analyses of endometrial cancer cells expressing exogenous PTEN. Cancer Res. 2001, 61, 3741–3749.
- Salemi, M.; Mazzetti, S.; de Leonardis, M.; Giampietro, F.; Medici, V.; Poloni, T.E.; Cannarella, R.; Giaccone, G.; Pezzoli, G.; Cappelletti, G.; et al. Poly (Adp-Ribose) Polymerase 1 and Parkinson’s Disease: A Study in Post-Mortem Human Brain. Neurochem. Int. 2021, 144, 104978.
- Salemi, M.; Galia, A.; Fraggetta, F.; La Corte, C.; Pepe, P.; La Vignera, S.; Improta, G.; Bosco, P.; Calogero, A. Poly (ADP-ribose) polymerase 1 protein expression in normal and neoplastic prostatic tissue. Eur. J. Histochem. 2013, 57, e13.
- Feng, Y.-C.A.; Cho, K.; Lindstrom, S.; Kraft, P.; Cormack, J.; Liang, L.; Driver, J.A. Investigating the genetic relationship between Alzheimer’s disease and cancer using GWAS summary statistics. Hum. Genet. 2017, 136, 1341–1351.
- Checler, F.; da Costa, C.A. P53 in Neurodegenerative Diseases and Brain Cancers. Pharmacol. Ther. 2014, 142, 99–113.
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell. Mol. Life Sci. 2020, 77, 2659–2680.
- Ariga, H. Common Mechanisms of Onset of Cancer and Neurodegenerative Diseases. Biol. Pharm. Bull. 2015, 38, 795–808.
- Fulda, S. Modulation of mitochondrial apoptosis by PI3K inhibitors. Mitochondrion 2013, 13, 195–198.
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial Energy Metabolism and Redox Signaling in Brain Aging and Neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371.
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517.
- Nagy, A.; Eder, K.; Selak, M.A.; Kalman, B. Mitochondrial energy metabolism and apoptosis regulation in glioblastoma. Brain Res. 2015, 1595, 127–142.
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis Via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top Med. Chem. 2020, 20, 524–539.
- Cai, Z.; Yan, L.-J.; Ratka, A. Telomere Shortening and Alzheimer’s Disease. Neuromol. Med. 2013, 15, 25–48.
- Panossian, L.A.; Porter, V.R.; Valenzuela, H.F.; Zhu, X.; Reback, E.; Masterman, D.; Cummings, J.L.; Effros, R.B. Telomere Shortening in T Cells Correlates with Alzheimer’s Disease Status. Neurobiol. Aging 2003, 24, 77–84.
- Depinho, R.A. The age of cancer. Nat. Cell Biol. 2000, 408, 248–254.
- Kim, N.; Piatyszek, M.; Prowse, K.; Harley, C.; West, M.; Ho, P.; Coviello, G.; Wright, W.; Weinrich, S.; Shay, J. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015.
- Clevers, H. Wnt/Beta-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480.
- Yao, H.; Sun, L.; Lian, J.; Zhang, M.; Liu, D. Correlation of Alzheimer’s Disease with Wnt Signaling Pathway and Neural Stem Cells. In Proceedings of the 4th International Conference on Mechatronics, Materials, Chemistry and Computer Engineering, Xi’an, China, 12–13 December 2015; pp. 119–122.
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nat. Cell Biol. 2005, 434, 843–850.
- Serafino, A.; Sferrazza, G.; Baldeschi, A.C.; Nicotera, G.; Andreola, F.; Pittaluga, E.; Pierimarchi, P. Developing drugs that target the Wnt pathway: Recent approaches in cancer and neurodegenerative diseases. Expert Opin. Drug Discov. 2017, 12, 169–186.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840.
- Houck, A.L.; Hernández, F.; Ávila, J. A Simple Model to Study Tau Pathology. J. Exp. Neurosci. 2016, 10, 31–38.
- Neckers, L. Heat shock protein 90: The cancer chaperone. J. Biosci. 2007, 32, 517–530.
- Frezza, M.; Schmitt, S.; Dou, Q.P. Targeting the ubiquitin-proteasome pathway: An emerging concept in cancer therapy. Curr. Top. Med. Chem. 2011, 11, 2888–2905.
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent Decrease in Global DNA Methylation and Hydroxymethylation in the Hippocampus of Alzheimer’s Disease Patients. Neurobiol. Aging 2013, 34, 2091–2099.
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; Laurent, G.S., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a Noncoding Rna Is Elevated in Alzheimer’s Disease and Drives Rapid Feed-Forward Regulation of Beta-Secretase. Nat. Med. 2008, 14, 723–730.
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222.
- Zhao, Y.; Jaber, V.; Alexandrov, P.N.; Vergallo, A.; Lista, S.; Hampel, H.; Lukiw, W.J. Microrna-Based Biomarkers in Alzheimer’s Disease (Ad). Front. Neurosci. 2020, 14, 585432.
- Vergallo, A.; Lista, S.; Zhao, Y.; Lemercier, P.; Teipel, S.J.; Potier, M.C.; Habert, M.O.; Dubois, B.; Lukiw, W.J.; Hampel, H.; et al. Mirna-15b and Mirna-125b Are Associated with Regional Abeta-Pet and Fdg-Pet Uptake in Cognitively Normal Individuals with Subjective Memory Complaints. Transl. Psychiatry 2021, 11, 78.
- Du, L.; Pertsemlidis, A. Cancer and neurodegenerative disorders: Pathogenic convergence through microRNA regulation. J. Mol. Cell Biol. 2011, 3, 176–180.
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol. Biol. 2017, 1509, 1–10.
This entry is offline, you can click here to edit this entry!