Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Applied
Over the past several decades, photopolymerization has become an active research field, and the ongoing efforts to develop new photoinitiating systems are supported by the different applications in which this polymerization technique is involved—including dentistry, 3D and 4D printing, adhesives, and laser writing.
- chalcone
- ketone
- photopolymerization
- photosensitizers
- Claisen-Schmidt condensation
1. Introduction
Polymerization consists of converting a liquid resin into a solid, and different approaches can be used to obtain this result. As the most popular approach, the polymerization can be instigated by heat, and for this purpose, various thermal polymerization techniques have been developed over the years—such as ring-opening polymerization (ROP) [1,2], reversible addition–fragmentation chain-transfer (RAFT) polymerization [3], and nitroxide-mediated polymerization (NMP) [4,5,6]. Parallel to this, light can also be used to generate initiating species. While, historically, photopolymerization was mostly based on UV photoinitiating systems, UV light is now the focus of numerous safety concerns, such that a great deal of effort is now being devoted to developing visible light photoinitiating systems offering safer working conditions for the operator (no skin or eye damage) [7,8,9,10,11,12,13,14,15,16]. Furthermore, improved light penetration can be achieved in the visible range compared to that obtained with UV light, as shown in Figure 1. Indeed, if light penetration remains limited in the UV range (600 µm), a major improvement can be achieved with visible light, which can range between 4 mm and 5 cm, depending on the irradiation wavelength [17]. As a result of this, the scope of application of photopolymerization has been totally revolutionized, since the use of near-infrared light now allows for the polymerization of thick and filled samples, which is not achievable with UV photoinitiating systems [18,19,20]. The development of visible light photoinitiating systems is also supported by the easy access to cheap, compact, lightweight, and energy-saving irradiation sources such as light-emitting diodes (LEDs) [21,22]. Faced with this easy availability of LEDs with tunable irradiation wavelengths, the demand for photopolymerizable resins activable at these different wavelengths has similarly increased. In particular, numerous photoinitiating systems activable at 405 nm have been developed over the last several years—this wavelength being the wavelength currently in use for 3D printers [23,24,25,26,27,28]. Interest in photopolymerization is also supported by the different advantages this polymerization technique offers compared to traditional thermal polymerization, which can only be realized in solution. Thus, photopolymerization can be carried out in solvent-free conditions so that the release of volatile organic compounds (VOCs) can be advantageously avoided [29,30,31,32]. Natural compounds, photoinitiators, and monomers issued from renewable resources can also be used to elaborate photoinitiating systems and polymers, addressing the environmental impact and the toxicity issues raised by photopolymerization, and by polymerization more generally [33]. A spatial and a temporal control can also be obtained, meaning that the polymerization occurs only during the time the light is switched on, and only in the irradiated area (see Figure 2) [34,35]. The polymerization process can also be extremely fast, since it can be ended within a few seconds. This specificity is notably used advantageously with photopolymerizable glues and dental adhesives.
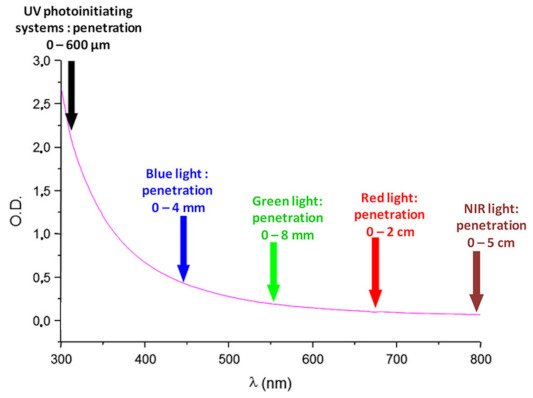
Figure 1. Light penetration in polystyrene latex with an average diameter of 112 nm. Reprinted with permission from Bonardi et al. [17]. Copyright 2018 American Chemical Society.
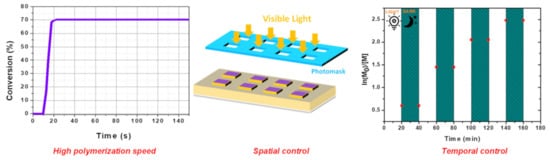
Figure 2. The different advantages of photopolymerization compared to traditional thermal polymerization.
Considering that visible light photopolymerization can be activated between 400 and 800 nm, numerous dyes absorbing in the visible range have been proposed, as exemplified with acridine-1,8-diones [36,37,38], carbazoles [39,40,41,42,43,44], pyrenes [45,46,47,48,49,50], iridium complexes, [51,52,53,54,55,56,57,58,59], copper complexes [60,61,62,63,64,65,66,67,68,69,70], squaraines [71,72,73], camphorquinones [74,75], perylenes [76,77,78], iodonium salts [79,80,81], benzophenones [82,83,84,85,86,87], cyanines [88,89], diketopyrrolopyrroles [90,91,92], helicenes [93,94], naphthalimides [95,96,97,98,99,100,101,102,103,104,105,106,107], chalcones [108,109,110,111,112,113,114], iron complexes [115,116,117,118,119,120], chromones [121,122,123], thioxanthones [124,125,126,127], dihydroanthraquinones [128], porphyrins [129,130], zinc complexes [131], acridones [132,133], push–pull dyes [134,135,136,137,138,139,140,141,142,143,144,145], phenothiazines [146], coumarins [147,148,149,150,151,152,153], flavones [154], 2,3-diphenylquinoxaline derivatives [155], and cyclohexanones [156,157,158,159]. With the aim of generating initiating species, two distinct families of photoinitiators can be distinguished: The first family, type I photoinitiators, consists of molecules that can be photochemically cleaved upon excitation. The advantage of this strategy is that only a single component is necessary to generate the initiating radicals, so the migratability of potential side products within the polymer is considerably reduced. As shown in Figure 3 with 2,2-dimethoxy-1,2-diphenylethan-1-one, upon photoexcitation, a methoxybenzyl and a benzoyl radical are simultaneously formed, improving the efficiency of the initiating step. Additionally, the two radicals can be connected to the polymer chain under growth so that no migratable residue remains within the polymer network, addressing the potential toxicity issue of the photoinitiating systems. However, while this approach is appealing, the availability of visible Type I photoinitiators remains limited, and most of the benchmark Type I photoinitiators are UV photoinitiators [160,161,162]. As a drawback, Type I photoinitiators are irreversibly consumed during the polymerization process, and so the concentration of radicals irreversibly decreases over time. Conversely, Type II photoinitiators are typically dyes absorbing in the visible range, which act as photosensitizers for UV photoinitiators. Upon photoinduced electron transfer from the excited photosensitizer towards the UV photoinitiator, initiating radicals can be generated [163]. As the most widely used UV photoinitiators, onium salts, and notably iodonium salts, which are easily accessible from various commercial sources can be cited as relevant examples [164,165,166,167]. Considering that dyes act as photosensitizers for UV photoinitiators, two-component or three-component photoinitiating systems are typically developed with Type II photoinitiators.
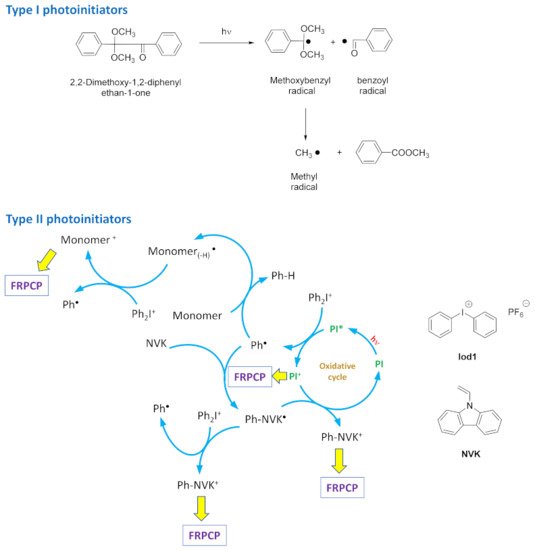
Figure 3. The two families of photoinitiators that have been developed in order to efficiently generate initiating radicals.
As shown in Figure 3, upon excitation of the photoinitiator with a light of an appropriate wavelength, a photoinduced electron transfer in the excited state can occur with the iodonium salt, generating phenyl radicals Ph●. These radicals can typically initiate the free-radical polymerization of acrylates. However, in these conditions, the consumption of the photosensitizer is irreversible, affecting the efficiency of the system. This drawback can be addressed by the addition of a third component—generally, a sacrificial amine that will be in charge of reducing the oxidized photosensitizer, and which can be introduced to the photocurable resin. If N-vinylcarbazole (NVK) is used, this carbazole can react with the phenyl radical Ph●, generating Ph–NVK●, which is a radical more reactive than the initial Ph● [168]. By reacting with the oxidized photosensitizer and regenerating the photosensitizer at its initial redox state, Ph–NVK● can be converted into a Ph–NVK+ cation, capable of initiating the cationic polymerization of epoxides by means of free-radical-promoted cationic polymerization (FRPCP). Therefore, using these three-component systems, the concomitant polymerization of acrylates and epoxides can be simultaneously obtained, enabling access to interpenetrated polymer networks (IPN) [169,170,171,172]. The photoinitiating systems are also catalytic if three-component systems are used, the regeneration of photoinitiators enabling the system to drastically reduce its content [173,174,175]. Considering that the photosensitizer is the key element of these two- and three-component photoinitiating systems, numerous structures have been examined. In this field, chalcones are dyes that can be naturally found in numerous vegetables and flowers [176,177,178]. Chalcones can also be easily obtained via a Claisen-Schmidt condensation. Considering their ease of synthesis, their strong absorption in the visible range, and their well-established biological activities, chalcones were investigated for applications ranging from medicine [179] to solar cell applications [180,181], organic light-emitting diodes [182], and organogels [183]. Among chalcones, bis-chalcones—which can be obtained via a Claisen-Schmidt condensation of aldehydes with cyclic aliphatic ketones in basic conditions—have been less studied in the literature than mono-chalcones [109,184]. Moreover, these structures remain of interest, especially for photopolymerization. Indeed, by increasing the molecular weight of photoinitiators, their migratability within the polymer network can be drastically reduced. These dyes also possess an extended conjugation compared to mono-chalcones; thus, these photosensitizers can transfer an electron towards an electron acceptor more easily in the excited state.
2. The Different Synthetic Routes to bis-Chalcones
Bis-chalcones have recently been studied as photoinitiators of polymerization, and three different strategies have been developed to provide access to these structures. Notably, chalcones can be easily obtained by means of a Claisen-Schmidt condensation between an aldehyde and an acetophenone, in accordance with the reaction depicted in Scheme 1 [186,187,188,189,190,191,192,193,194,195]. No major differences can be found from the synthetic viewpoint between mono- and bis-chalcones, except that two equivalents of aldehydes have to be used in the case of bis-chalcones, comprising a central cyclic ketone. In addition to this first strategy based on cyclic ketones, a second approach can consist of connecting two mono-chalcones together. In this aim, two connected aldehydes or two connected acetophenones can be used to form bis-chalcones.
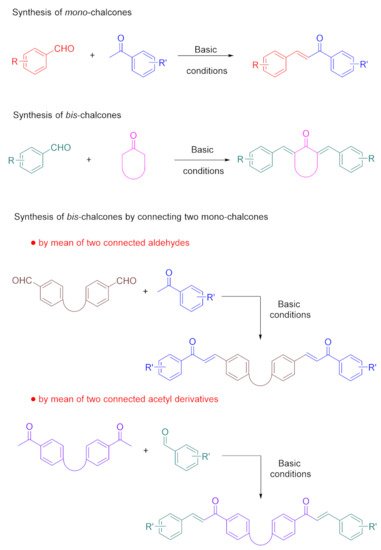
Scheme 1. Synthetic routes to mono- and bis-chalcones; and bis-chalcones obtained by connecting two mono-chalcones.
Based on the synthetic approach used to provide access to these structures, numerous modifications of the chalcone scaffold can be envisioned, such as a modification of the peripheral groups, the substitution pattern of the central cyclic aliphatic ketone, or the spacer introduced between the central core and the peripheral groups (see Figure 4). In the same spirit, bis-chalcones can be obtained via the condensation of acetophenones on bis-aldehydes. Aldehydes can also be condensed onto bis-acetophenones. Here, again, this strategy is highly versatile; since the spacer is used to connect the two chalcones, the substitution pattern of the chalcones can be easily tuned. Overall, the connection of two chalcones together enables a similar effect on the migratability of these macrophotoinitiators.
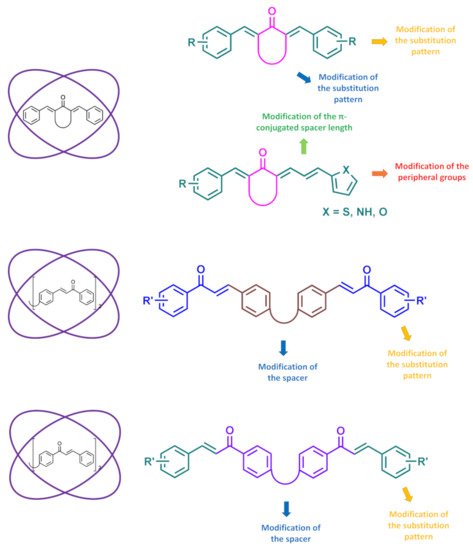
Figure 4. The different chemical modifications enabling the efficient tuning of the absorption spectra of bis-chalcones.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26113192
This entry is offline, you can click here to edit this entry!