ATM is among of the most critical initiators and coordinators of the DNA-damage response. ATM canonical and non-canonical signaling pathways involve hundreds of downstream targets that control many important cellular processes such as DNA damage repair, apoptosis, cell cycle arrest, metabolism, proliferation, oxidative sensing, among others. Of note, ATM is often considered a major tumor suppressor because of its ability to induce apoptosis and cell cycle arrest. However, in some advanced stage tumor cells, ATM signaling is increased and confers remarkable advantages for cancer cell survival, resistance to radiation and chemotherapy, biosynthesis, proliferation, and metastasis.
- ATM
- cancer
- DNA damage repair
- DNA damage response
- oxidative sensing
- autophagy
- hypoxia
1. Introduction
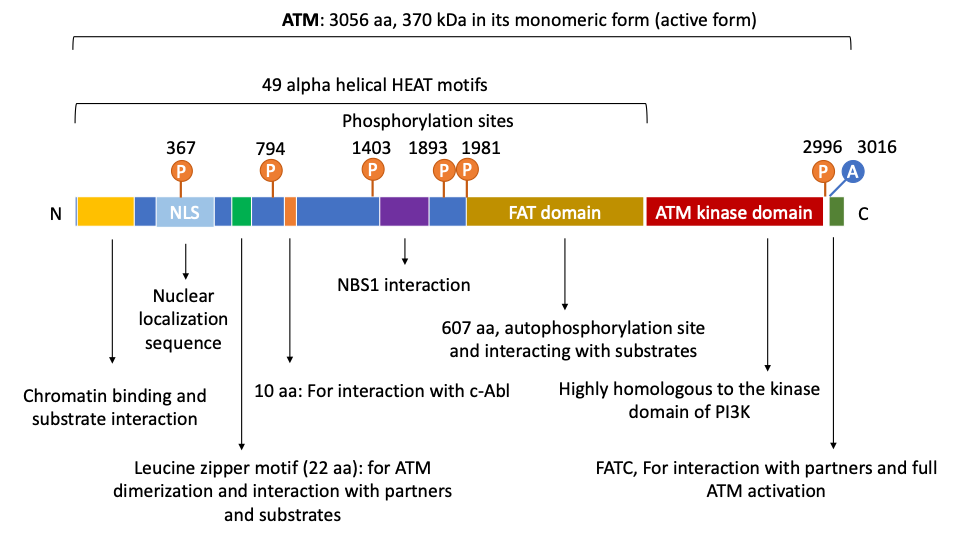
2. The Structure and Domain Mapping of ATM
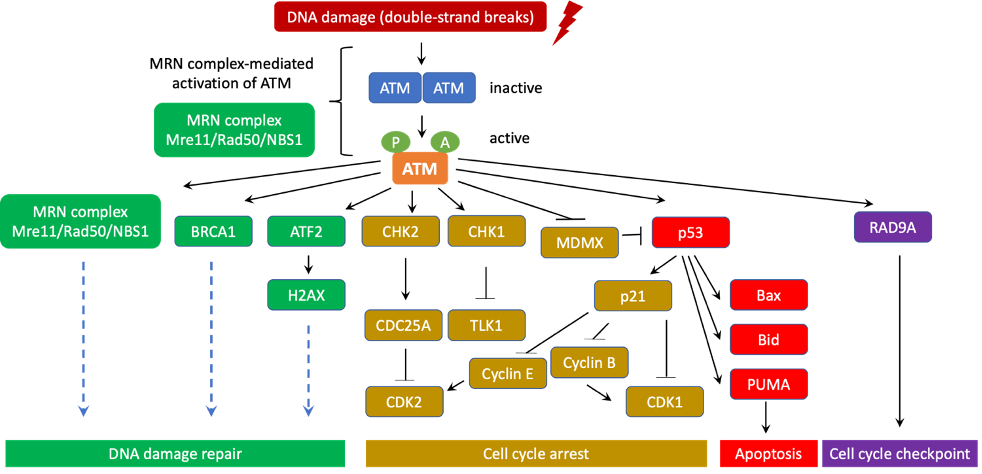
3. ATM’s role in DNA Damage Response and Ataxia Telangiectasia
This entry is adapted from the peer-reviewed paper 10.3390/genes12060845
References
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; A Tagle, D.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753.
- Abraham, R.T. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair 2004, 3, 883–887.
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738.
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008.
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196.
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–227.
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC Domains of PIKK Proteins Are Functionally Equivalent and Participate in the Tip60-dependent Activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746.
- Fernandes, N.; Sun, Y.; Chen, S.; Paul, P.; Shaw, R.J.; Cantley, L.C.; Price, B.D. DNA Damage-induced Association of ATM with Its Target Proteins Requires a Protein Interaction Domain in the N Terminus of ATM. J. Biol. Chem. 2005, 280, 15158–15164.
- Andrade, M.A.; Petosa, C.; I O’Donoghue, S.; Müller, C.W.; Bork, P. Comparison of ARM and HEAT protein repeats. J. Mol. Biol. 2001, 309, 1–18.
- Piazza, I.; Rutkowska, A.; Ori, A.; Walczak, M.; Metz, J.; Pelechano, V.; Beck, M.; Haering, C.H. Association of condensin with chromosomes depends on DNA binding by its HEAT-repeat subunits. Nat. Struct. Mol. Biol. 2014, 21, 560–568.
- Rubinson, E.H.; Gowda, A.S.P.; Spratt, T.E.; Gold, B.; Eichman, B.F. An Unprecedented Nucleic Acid Capture Mechanism for Excision of DNA Damage. Nature 2010, 468, 406–411.
- Dupré, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the Mre11–Rad50–Nbs1 complex. Nat. Struct. Mol. Biol. 2006, 13, 451–457.
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611.
- Kanu, N.; Behrens, A. ATMIN defines an NBS1-independent pathway of ATM signalling. EMBO J. 2007, 26, 2933–2941.
- Bowen, C.; Ju, J.-H.; Lee, J.-H.; Paull, T.T.; Gelmann, E.P. Functional activation of ATM by the prostate cancer suppressor NKX3.1. Cell Rep. 2013, 4, 516–529.
- Lavin, M.F. The Mre11 complex and ATM: A two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair 2004, 3, 1515–1520.
- Lavin, M.F.; Scott, S.; Gueven, N.; Kozlov, S.; Peng, C.; Chen, P. Functional consequences of sequence alterations in the ATM gene. DNA Repair 2004, 3, 1197–1205.
- Lee, J.H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554.
- Lee, J.H.; Paull, T.T. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science 2004, 304, 93–96.
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621.
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210.
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506.
- Berkovich, E.; Monnat, R.J., Jr.; Kastan, M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007, 9, 683–690.
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.-Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-Dependent and -Independent Dynamics of the Nuclear Phosphoproteome After DNA Damage. Sci. Signal. 2010, 3, rs3.
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166.
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583.
- Álvarez-Quilón, A.; Serrano-Benítez, A.; Lieberman, J.A.; Quintero, C.; Sánchez-Gutiérrez, D.; Escudero, L.M.; Cortés-Ledesma, F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 2014, 5, 3347.
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Gatti, R.A.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.G.J.; Lange, K.; et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature 1988, 336, 577–580.
- Taylor, A.M.; Byrd, P.J. Molecular pathology of ataxia telangiectasia. J. Clin. Pathol. 2005, 58, 1009–1015.
- Chun, H.H.; A Gatti, R. Ataxia–telangiectasia, an evolving phenotype. DNA Repair 2004, 3, 1187–1196.
- Renwick, A.; The Breast Cancer Susceptibility Collaboration (UK); Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875.