Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
A disturbed interaction between the gut microbiota and the mucosal immune system plays a pivotal role in the development of inflammatory bowel disease (IBD).
- microbial metabolites
- gut
- inflammation
1. Introduction
Inflammatory bowel disease (IBD) is an idiopathic disease affecting the gastrointestinal (GI) tract and can be divided into two main subcategories: Crohn’s disease (CD) and ulcerative colitis (UC). Both CD and UC lead to poor quality of life and psychological distress for patients, and produce significant pressure on healthcare systems by their relatively high morbidity. Genetic and environmental factors are known to increase the risk of IBD and may predispose certain individuals or populations to developing the disease. Prevalence of IBD has always been relatively high in Europe and North America, but is now also on the rise in industrializing countries in Asia, Africa, and South America [1].
Despite the lack of full understanding of the pathophysiology of IBD, the majority of available reports suggest a dysregulation between the intestinal microbiota and the host immune system (i.e., loss of immune tolerance) to be one of the underlying causes. The innate immune system in the intestinal mucosa responds to the microbiota and/or antigens by promoting inflammation, which recruits the adaptive immune system and leads to a more severe and long-lasting inflammatory state, as well as deterioration of the intestinal barrier integrity. The latter leads to translocation of microbiota and/or antigens into the mucosa, further exacerbating the mucosal inflammatory response, thereby creating a vicious circle [2,3].
Currently used pharmacological interventions are aimed at combatting the characteristic flareups of intestinal inflammation. The most effective drugs are corticosteroids and tumor necrosis factor (TNF) inhibitors. However, the former cannot be used for extended periods of time due to serious side effects (e.g., Cushing’s syndrome), and the latter has a significant amount of primary and secondary non-responders, along with serious side effects [4,5,6].
Fecal microbiota transplant (FMT) is another, experimental, form of IBD treatment. A recent meta-analysis found that 54% of IBD patients showed a clinical response to FMT, and 37% demonstrated clinical remission, while 29% suffered from adverse events [7]. Generally, the adverse events following FMT are mild and subside within 24 h, but more serious events, such as IBD flareups, infections, colectomy, pancreatitis, and death are also reported, although less frequently [8].
Despite the promising remission rates of this IBD treatment, which is still in its infancy, the main motive against FMT is that the treatment is considered to be a black box. The outcome and safety of the treatment is influenced by a myriad of factors (e.g., host genotype, specific type of microbiota imbalance, type and stage of IBD, route of administration, and factors related to the FMT donor), which remain obscure [8,9].
Considering the pivotal role of the gut microbiota in IBD, and that, ultimately, a major part of the communication between the gut microbiota and the host is based on chemical signaling, this review aims to examine gut microbial metabolites known to have anti-IBD effects. In order to positively implicate the role of microbial metabolism, only compounds proven to be produced by the gut microbiota have been taken into consideration. Furthermore, the metabolites discussed in this review originate from parental compounds found in common dietary sources (e.g., vegetables, fruits, and herbs), and have either been shown to improve colitis symptoms in vivo, affect signaling pathways involved in the pathophysiology of IBD in vitro, or both. The relevant data are summarized in Table 1.
Table 1. Overview of metabolites, bacterial species currently known to produce these metabolites, and experimental models used to assess anti-IBD effects.
| Microbial Metabolite | Parental Compound | Phylum | Species | Experimental Model | Ref. |
|---|---|---|---|---|---|
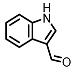 Indole-3-aldehyde (I3Al) |
Tryptophan | Firmicutes | Lactobacillus reuteri Lactobacillus murinus |
in vitro, in vivo |
[10,11,12] |
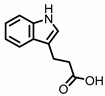 Indole-3-propionic acid (I3Pr) |
Tryptophan | Firmicutes | Peptostreptococcus russellii Peptostreptococcus anaerobius Peptostreptococcus asaccharolyticus Clostridium sporogenes Clostridium botulinum Clostridium caloritolerans Clostridium paraputrificum Clostridium cadaveris |
in vitro, in vivo |
[13,14,15,16,17,18] |
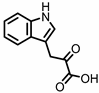 Indole-3-pyruvic acid (I3Py) |
Tryptophan | Firmicutes | Clostridium sporogenes | in vitro, in vivo |
[15,19] |
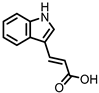 Indole-3-acrylic acid (I3Acr) |
Tryptophan | Firmicutes | Peptostreptococcus russellii Peptostreptococcus anaerobius Clostridium sporogenes |
in vitro | [15,20] |
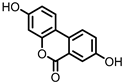 Urolithin A (UrA) |
Ellagic acid | Actinobacteria | Bifidobacterium pseudocatenulatum | in vitro, in vivo |
[21,22,23,24,25,26] |
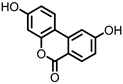 Isouroithin A (iUrA) |
Ellagic acid | Actinobacteria | Ellagibacter isourolithinifaciens | in vitro | [25,27,28] |
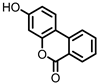 Urolithin B (UrB) |
Ellagic acid | Actinobacteria | Bifidobacterium pseudocatenulatum | in vitro | [21,24,25] |
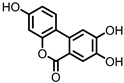 Urolithin C (UrC) |
Ellagic acid | Actinobacteria | Gordonibacter urolithinfaciens Gordonibacter pamelaeae |
in vitro | [24,29,30] |
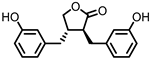 Enterolactone (EL) 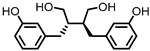 Enterodiol (ED) |
Lignans | Firmicutes | Lactobacillus gasseri Lactobacillus salivarius Clostridium scindens Lactonifactor longoviformis Peptostreptococcus productus |
in vitro | [31,32,33,34,35,36,37,38,39,40] |
| Actinobacteria | Bifidobacterium bifidum Bifidobacterium catenulatum Bifidobacterium pseudolongum Bifidobacterium adolescentis Eggerthella lenta |
||||
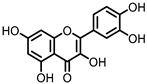 Quercetin |
Quercitrin | Fusobacteria | Fusobacterium K-60 | in vitro, in vivo |
[41,42,43,44] |
| Rutin | Firmicutes | Enterococcus avium Lactobacillus acidophilus Lactobacillus plantarum Lachnoclostridium spp. Eisenbergiella spp. Blautia sp. |
in vitro, in vivo |
[45,46,47,48,49,50,51,52,53] | |
| Actinobacteria | Bifidobacterium dentium | ||||
| Bacteroidetes | Bacteroides uniformis Bacteroides ovatus Parabacteroides distasonis |
||||
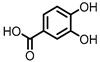 Protocatechuic acid (PCA)/3,4-dihydroxybenzoic acid 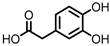 3,4-dihydroxyphenylacetic acid (DHPA) 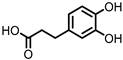 3,4-dihydroxyphenylpropionic acid (DHPP) |
Flavonols Flavan-3-ols Flavones Anthocyanins |
Firmicutes | Eubacterium oxidoreducens Eubacterium ramulus Enterococcus casseliflavus Flavonifractor plautii Catenibacillus scindens Butyrivibrio spp. |
in vitro, in vivo |
[54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69] |
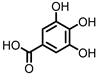 Gallic acid (GA)/3,4,5-trihydroxybenzoic acid |
Anthocyanins | Firmicutes | Lactobacillus plantarum Lactobacillus casei |
in vitro, in vivo |
[70,71,72,73,74,75,76,77] |
| Actinobacteria | Bifidobacterium lactis | ||||
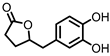 3,4-dihydroxyphenyl-γ-valeric lactone (DHPVL) |
Flavan-3-ols Proanthocyanins |
Firmicutes | Lactobacillus plantarum Clostridium coccoides Flavonifractor plautii |
in vitro | [54,55,58,59,60,63,78,79,80] |
| Actinobacteria | Eggerthella lenta Eggerthella sp. |
||||
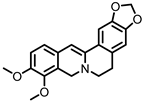 Dihydroberberine |
Berberine | Firmicutes | Enterococcus faecium Enterococcus faecalis Staphylococcus aureus Staphylococcus epidermis |
in vitro a, in vivo | [81,82,83,84,85,86,87] |
| Proteobacteria | Escherichia coli Enterobacter cloacae Klebsiella pneumoniae |
||||
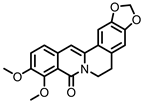 Oxyberberine |
Berberine | Firmicutes | Lactobacillus acidophilus Streptococcus aureus |
in vivo | [88] |
| Actinobacteria | Bifidobacterium longum | ||||
| Proteobacteria | Escherichia coli Pseudomonas aeruginosa |
||||
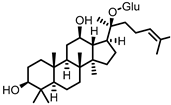 Compound K (CK) |
Ginsenoside Rb1 | Firmicutes | Eubacterium | in vitro, in vivo |
[89,90,91,92,93,94] |
| Actinobacteria | Bifidobacterium | ||||
| Bacteroidetes | Bacteroides | ||||
| Fusobacteria | Fusobacterium |
Due to intrinsic differences in the interindividual dietary and microbiota compositions, especially the disturbed microbiota of IBD patients, such metabolites may not be produced universally. Identifying these metabolites can help to overcome such intrinsic differences, and, ideally, helps making gut health less dependent on changes in the microbiota composition.
2. Indoles
Indole derivatives (Figure 1) are mainly produced by Lactobacilli, Clostridia, Peptostreptococci, Bifidobacteria, and Bacteroides (Table 1), as metabolites of the amino acid tryptophan (Trp) [95]. Gut microbial Trp metabolites are often found to be agonists of the aryl hydrocarbon receptor (AHR), of which lower levels are observed in IBD patients, compared to healthy subjects [96]. IBD symptoms and pro-inflammatory cytokine levels were found to be greater in AHR knockouts in murine models of dextran sodium sulfate (DSS)-induced colitis [97]. Other AHR ligands are known to reduce colitis symptoms [96,98].
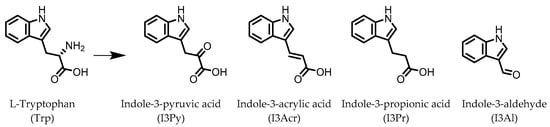
Figure 1. Structures of L-tryptophan and several indole metabolites produced by the gut microbiota.
AHR activation by the gut microbial Trp metabolite indole-3-aldehyde (I3Al) was shown to stimulate mucosal lymphocytes to secrete interleukin 22 (IL-22), an anti-inflammatory cytokine known to play an important role in protecting mice from developing IBD [99]. Increased IL-22 secretion causes signal transducer and activator of transcription 3 (STAT3) phosphorylation, which ultimately leads to faster proliferation of intestinal epithelial cells (IECs), contributing to the recovery of damaged intestinal mucosa following DSS-induced colitis [10].
Indole-3-propionic acid (I3Pr) also activates the AHR receptor, which induced IL-10 receptor expression in cultured IECs. Oral administration of I3Pr was shown to improve DSS-induced murine colitis symptoms, which was attributed to increased signaling of the anti-inflammatory cytokine IL-10, due to higher expression of IL-10 receptors [13].
Additionally, I3Pr was found to act as a ligand for the pregnane X receptor (PXR) in vivo, and led to lower TNF-α levels together with higher levels of mRNA coding for tight junction proteins, thus contributing to intestinal integrity. With the help of knockout experiments, it was determined that activation of PXR modulates Toll-like receptor 4 (TLR4) signaling, which is known to activate nuclear factor κB (NF-κB), a pro-inflammatory transcription factor. Accordingly, oral administration of I3Pr could activate PXR in the colon, which prevents lipopolysaccharide (LPS)-induced inflammation via modulation of TLR4, thereby preserving the intestinal integrity [14].
Administration of indole-3-pyruvic acid (I3Py) to mice with CD4+ T cell-induced colitis led to an increase in the amount of IL-10-producing T cells, while the number of Th1 cells in the mucosa was decreased, resulting in a reduction in colitis symptoms [19].
In a co-culture of murine-derived colonic spheroids and murine bone marrow-derived macrophages (BMDMs), indole-3-acrylic acid (I3Acr) promoted IL-10 secretion while suppressing TNF-α production upon stimulation with LPS, via activation of AHR. This stimulated the expression of the mucin protein coding gene, Muc2, which may help to protect the intestinal epithelium. When human peripheral blood mononuclear cells (PBMCs) were treated with I3Acr, a reduction in IL-1β and IL-6 was observed, upon LPS stimulation. Moreover, not only was AHR activation reproduced in the human cell line, activation of the anti-inflammatory Nrf2–ARE pathway was observed. Using these human PBMCs in the co-culture, I3Acr treatment promoted important anti-inflammatory and anti-oxidant effects, by upregulating Nrf2- and AHR-pathway target genes and genes related to the biosynthesis glutathione (GSH), an important anti-oxidant that protects cells from oxidative stress [20].
3. Urolithins
Urolithins are gut microbial metabolites of ellagic acid, a hydrolysis product of ellagitannins (Figure 2). Both ellagic acid and ellagitannins are naturally found in various fruits, nuts, and seeds (e.g., pomegranate, raspberry, strawberry, almond, and walnut) [100]. Several members of the Actinobacteria (Table 1) have been found to metabolize ellagic acid into particular urolithins, which differ by the number and the positions of hydroxyl groups. For example, Gordonibacter urolithinfaciens and Gordonibacter pamelaeae are able to produce urolithin C (UrC), but are not capable of further dehydroxylation [29,30]. Urolithin A (UrA) and urolithin B (UrB) are produced by Bifidobacterium pseudocatenulatum, whereas isourolithin A (iUrA) is produced by Ellagibacter isourolithinifaciens [21,27,28].
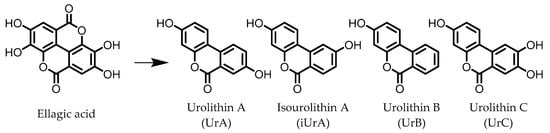
Figure 2. Structures of ellagic acid and several urolithins produced by the gut microbiota.
A comparison between the effects of pomegranate extract (PE) and UrA on DSS-induced colitis in rats showed that both were able to decrease levels of the pro-inflammatory mediators nitric oxide (NO) and prostaglandin E2 (PGE2) in colonic mucosa, by downregulating the enzymes responsible for their production: inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2), and prostaglandin E synthase (PTGES). However, only in the case of UrA administration was the colonic architecture protected. Additionally, UrA was able to significantly downregulate the pro-inflammatory cytokines IL-1β and IL-4, and cluster of differentiation 40 (CD40), a receptor protein involved in immune and inflammatory signaling pathways [22].
It was also observed that less UrA was produced from PE in colitic rats compared to healthy rats, suggesting that UrA production from gut microbiota, which might be absent in inflammation, plays a protective role against colitis. During colitis, UrA itself had to be administered in order to benefit from the anti-inflammatory effects. Another protective effect of UrA might be via an observed increase in the abundance of Lactobacilli, Bifidobacteria, and Clostridia taxa, which have been shown to prevent inflammation in IECs in response to pathogenic Enterobacteria [101]. Moreover, an increase in E. coli, observed after DSS treatment, was found to be lower in the rats that received UrA [22].
Several in vitro studies have been performed in an attempt to reveal a more detailed mechanism explaining the anti-inflammatory actions of UrA. The production of pro-inflammatory mediators was strongly reduced by UrA in LPS-stimulated RAW264 macrophages. UrA was found to inhibit the phosphorylation of protein kinase B (Akt) and c-Jun, effectively suppressing the pro-inflammatory PI3-K/Akt/NF-κB and JNK/AP-1 signaling pathways. This meant the downstream production of pro-inflammatory mediators (TNF-α, IL-6, and NO) was also suppressed. Notably, UrA appeared to also inhibit NADPH oxidase (NOX), which is largely responsible for production of reactive oxygen species (ROS) in activated macrophages, presenting another possible mechanism for inhibiting the activation of the pro-inflammatory transcription factors NF-κB and AP-1 [23].
iUrA, UrB, and UrC also display anti-inflammatory effects in LPS-stimulated RAW264.7 macrophages, although the effects are inferior to UrA. The urolithins were shown to decrease the DNA-binding activity of the NF-κB p50 subunit, as well as the nuclear translocation of the p65 subunit, resulting in lower levels of TNF-α, IL-1β, IL-6, iNOS, and NO [24,25]. Additionally, UrA has been shown to promote anti-inflammatory effects in human macrophages and neutrophils, which was attributed to an observed induction of extracellular signal-regulated kinase 1 and 2 (ERK1/2) phosphorylation [25].
Besides the anti-inflammatory properties and modulation of the microbiota, UrA can also improve gut health by enhancing the intestinal barrier function. UrA was shown to activate AHR and Nrf2, which leads to the upregulation of the tight junction proteins claudin 4, occludin, and zonula occludens-1 (ZO-1). Treatment with UrA decreased gut permeability in mice with 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis, and reduced both local and systemic inflammation. When UrA was administered prior to TNBS-administration, the development of colitis was prevented. Finally, chronic and acute DSS-induced colitis were ameliorated by UrA treatment [26].
This entry is adapted from the peer-reviewed paper 10.3390/ph14060506
This entry is offline, you can click here to edit this entry!