ATM is among of the most critical initiators and coordinators of the DNA-damage response. ATM canonical and non-canonical signaling pathways involve hundreds of downstream targets that control many important cellular processes such as DNA damage repair, apoptosis, cell cycle arrest, metabolism, proliferation, oxidative sensing, among others. Of note, ATM is often considered a major tumor suppressor because of its ability to induce apoptosis and cell cycle arrest. However, in some advanced stage tumor cells, ATM signaling is increased and confers remarkable advantages for cancer cell survival, resistance to radiation and chemotherapy, biosynthesis, proliferation, and metastasis.
- ATM
- cancer
- DNA damage repair
- DNA damage response
- oxidative sensing
- autophagy
- hypoxia
1. Introduction
2. The Structure and Domain Mapping of ATM
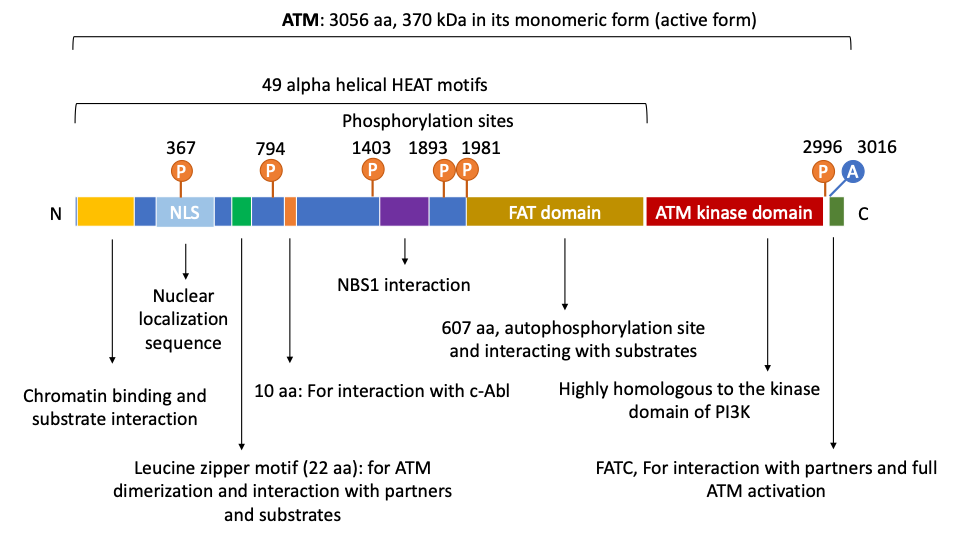
3. ATM’s role in DNA Damage Response and Ataxia Telangiectasia
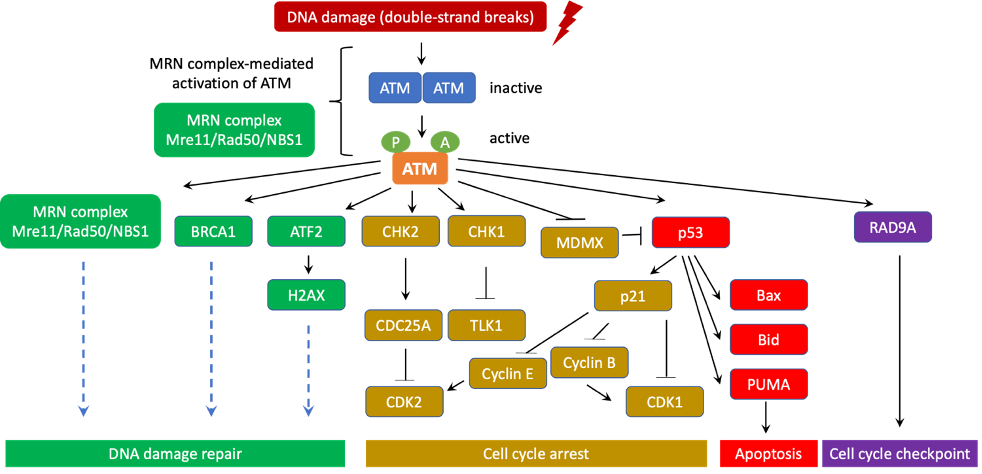
4. ATM Signaling Pathways
ATM signaling pathways could be classified into the canonical pathway and a diverse array of non-canonical pathways. In the canonical pathway, ATM collaborates with the MRN complex (Mre11/Rad50/NBS1) to activate the cellular DNA-damage response. ATM also uses a number of other non-canonical pathways to respond to other types of cellular stresses [34].
Similar to other PI3K family members, ATM kinase activity is strictly autoinhibited during its resting state (dimers or polymers) and only active when ATM binds to its partners. In response to DNA double-strand breaks (DSBs), ATM canonical pathway is induced, which involves the disassociation of ATM from dimers to monomers, activation and recruitment of ATM monomers to the DNA damage sites. It has been well documented that the MRN complex is required for ATM activation and recruitment to DSB locations. Indeed, the NBS1 subunit of the MRN complex directly binds to ATM and NBS1 ubiquitination promotes ATM recruitment to DSBs [18,20,22,35]. Subsequently, ATM phosphorylates a number of downstream targets as CHK1, CHK2, p53, BRCA1, ATF2, among others to stimulate DNA damage repair machinery [21,24].
In human, autophosphorylation at Ser1981 is often considered as a marker for ATM activation, which often occurs quickly after ionizing radiation [22]. In the absence of the MRN complex, ATM Ser1981 is not phosphorylated but ATM is still able to phosphorylate the downstream histone protein H2AX after radiation [12]. It is postulated that while phosphorylation at Ser1981 is not required for ATM function, this phosphorylation is critical to retain ATM at the DSBs [23,36]. Subsequent to H2AX phosphorylation, DNA repair complexes containing effectors, polymerases, among others are recruited to the DSB site. For instance, after being phosphorylated by ATM, MDC1 recruits RNF8, RNF168 [37–39], which in turn monoubiquitinates histone H2AX and H2A on Lys13 and Lys15 to prepare them for additional poly-ubiquitination reactions. These posttranslational protein modifications are necessary to recruit additional effector repair proteins [40]. It has been shown that continuous ATM activation is critical to maintain the damage foci for sustaining effective DNA damage repair [41]. It is important to point out that ATM-mediated signaling for DNA damage repair is tightly regulated by several feedback mechanisms to ensure proper and effective repair efficiency. For instance, it has been shown that due to the feedback of p53, ATM activation occurs in a pulse manner [42].
Chromatin decompaction and relaxation at DSB sites are essential for effective repair and ATM activation. In fact, ATM phosphorylates RNF20/RNF40 to facilitate histone H2B ubiquitination. ATM-mediated phosphorylation of KAP1 also decreases KAP1′s binding to CHD3, a chromodomain protein. These two events result in chromatin decompaction [43–45]. Of note, histone deacetylase inhibitors have been shown to activate ATM [22]. The histone acetyltransferase complex TIP60 is necessary for ATM activation after irradiation [46]. Interestingly, by increasing H3K14 acetylation and loosening chromatin structure, HMGN1 significantly elevates the amount of chromatin-bound ATM at DSB sites [47]. In contrast, to silence transcription at and near DNA damage sites, ATM promotes Histone H2A K119 mono-ubiquitination by BMI1 [48–50].
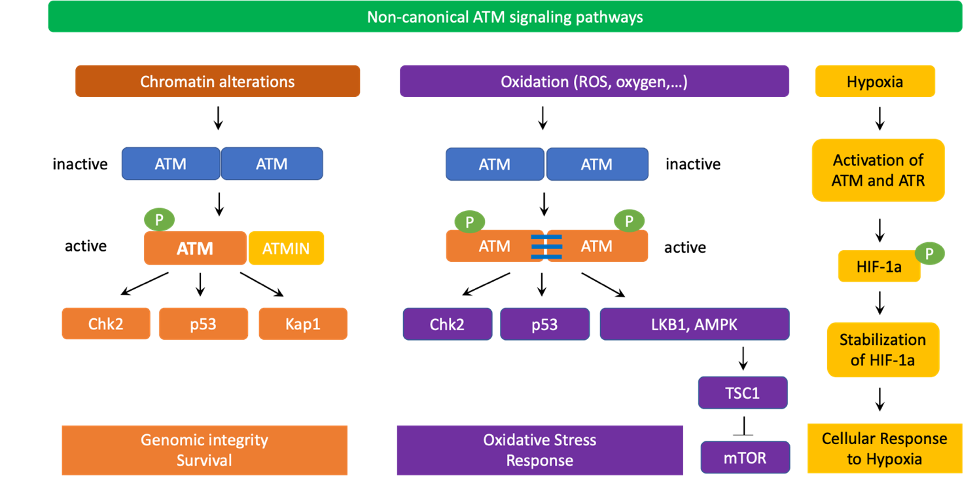
Figure 3. ATM non-canonical signaling pathways. Chromatin alterations induced by chloroquine or hypotonic stress could trigger ATM activation without requiring the MRN complex. The active monomer of ATM interacts with ATMIN (ATM interacting protein) to transduce downstream signaling to CHK2, p53, KAP1, and other substrates to promote genomic integrity and survival. ATM also serves as an important redox sensor. After being activated by ROS or oxidation, ATM homodimers establish disulfide bonds to maintain their dimer conformations. This activation process also does not require the MRN complex. Similar to active ATM monomers, induced ATM dimers are phosphorylated at Ser1981 but this posttranslational modification is not required for phosphorylation of downstream targets such as CHK2 at Thr68 or p53 at Ser18. Oxidation-activated ATM dimers then phosphorylate LKB1 and AMPK to turn on TSC2 and block mTOR signaling, thereby decreasing ROS levels. ATM can also be induced by severe hypoxia in a ROS-independent manner. In severe hypoxia, the function of ribonucleotide reductase is inhibited, causing the depletion of deoxynucleoside triphosphates (dNTPs) as well as replication stress. As a result, ATM and ATR are activated due to severe hypoxia. ATM and ATR then phosphorylate and stabilize HIF1α to enable cell survival under hypoxic conditions.
In non-canonical signaling pathways, ATM can also be activated by chromatin changes caused by chloroquine, hypotonic cellular stress, among others [22,51] (Figure 3). Interestingly, the MRN complex, especially NBS1, is not required for ATM non-canonical activation and signaling. Instead, ATMIN (ATM interacting protein) is required for ATM activation in these scenarios. Interestingly, ATMIN binds to ATM using interacting domains that are similar to those of NBS1 [52]. ATMIN is also partially required for ATM-mediated phosphorylation of downstream targets in non-canonical ATM pathways [53]. Furthermore, ATM also cross-talks with ATR for activation in response to UV radiation [54]. Importantly, in non-replicating cells, ATM can be activated by R-loops at transcription-blocking lesions. In fact, in 2015, Tresini et al., discovered that RNA polymerase pausing at the DNA damaged sites led to spliceosome displacement and the formation of R-loops, which subsequently activated ATM. R-loop-mediated activation of ATM prevented further spliceosome organization and increased genome-wide ultraviolet-irradiation-induced alternative splicing [55].
ATM is also involved in sensing oxidative stress. In this case, ATM dimers are oxidized and establish disulphide bonds between ATM monomers to create active ATM dimers and phosphorylate downstream targets. Exposure to oxygen at atmospheric levels or reactive oxygen species (ROS) activate ATM without requiring the MRN complex [56]. Active ATM dimers also carry phosphorylated Ser1981 but this phosphorylation is dispensable for ATM dimers’ function. Interestingly, after being induced by ROS, ATM dimers activate TSC2 by phosphorylating LKB1 and AMPK, thereby blocking mTOR signaling and reducing ROS production [57]. Importantly, ATM is activated by hypoxia and phosphorylates the transcription factor HIF1α, which leads to HIF1α stabilization and REDD1-mediated inhibition of mTOR [58]. Indeed, severe hypoxia impedes the activity of ribonucleotide reductase, leading to the depletion of dNTPs and subsequent replication stress. As a result, activation of ATM and ATR occurs. ATM and ATR then directly phosphorylate and stabilize HIF1α to promote cell survival under severe hypoxic conditions [59–62]. In 2017, Rezaeian et al. also showed that ATM indirectly induced HIF1α stability through H2AX phosphorylation in the cross talk with TRAF6-mediated H2AX ubiquitination [63]. As hypoxia and increased ROS stress frequently occur in solid tumors, ATM may promote cancer cell survival in some scenarios by stabilizing HIF1α and reducing ROS levels.
Recent studies additionally revealed a connection between ATM and autophagy [64–66] in response to nutrient deprivation, ROS, and DNA damage. Indeed, after being activated by oxidative stress or genotoxic agents, ATM suppresses mTORC1 while inducing autophagy [57,67,68]. ATM also increases the expression of ATG4C at the mRNA and protein levels, promoting autophagy in breast cancer stem cells [69]. Furthermore, ATM participates in regulating pexophagy in response to ROS [70,71]. Pexophagy is a catabolic process to selectively degrade peroxisomes by autophagy where autophagosomes engulf peroxisomes and fuse with lysosomes for subsequent degradation of peroxisomes. Pexophagy is critical to maintain cellular homeostasis by removing damaged or unnecessary peroxisomes. In the case of pexophagy, ATM is localized to peroxisomes through the PEX5 import receptor, which recognizes an SRL sequence on ATM’s C-terminus. As a result of ROS-mediated activation, ATM phosphorylates TSC2 to inhibit mTORC1, and ULK1 to promote autophagy. Furthermore, ATM directly phosphorylates PEX5 at Ser141. This phosphorylation subsequently leads to PEX5 ubiquitination at Lys209 and association with SQSTM1/p62 (an autophagy receptor) to trigger pexophagy [70,71]. In addition to genotoxic and oxidative stresses, ATM can be also activated by starvation and reactive nitrogen species [72]. Cytoplasmic ATM presence have been found in mitochondria peroxisomes, and endosomes, where the kinase exerts its regulatory functions in sensing oxidative stress and participate in the regulation of cellular metabolism and autophagy processes [73]. Some early evidence suggested that ATM helps maintain cellular homeostasis in response to DNA damage and ROS via the autophagy-senescence signaling axis [74]. Moreover, ATM has been documented to regulate mitophagy, a process to selectively remove mitochondria at the autophagolysosomes for turning over the dysfunctional or damaged mitochondria for adapting to physiological stress conditions, such as nutrient deprivation, hypoxia, DNA damage, among others. Recent evidences showed that ATM modulated Beclin-1 to control mitophagy [75,76] and mediated spermidine-induced mitophagy via regulating PINK expression and Parkin localization to mitochondria [77]. Together, these findings indicate an important role of ATM in sensing oxidative stress as well as regulating autophagy, mitophagy, and pexophagy to maintain cellular homeostasis.
5. ATM Signaling in Cancer
ATM’s major tumor suppressing mechanisms are inducing apoptosis and cell cycle arrest via activating p53, SIRT1, CHK1, CHK2, DBC1, RAIDD and other downstream targets [34]. Therefore, cancer cells can use different mechanisms to downregulate ATM. For instance, in breast cancer cells, ATM expression can be reduced due to miRNA-18a [78,79]. ATM activity could also be suppressed by the phosphatase WIP1, which directly dephosphorylates ATM and p53 [80].

Figure 4. The diverse roles of ATM in cancer cells. In some cancer cells that have developed mechanisms to evade apoptosis and cell cycle arrest, increase in ATM signaling may confers significant advantages for cancer cell survival, biosynthesis, proliferation, metastasis, as well as resistance to radiation and chemotherapy.
Interestingly, in some tumor cells, ATM signaling and function are upregulated. Perhaps, those cancer cells have developed mechanisms to escape ATM-induced cell cycle arrest and apoptosis. Indeed, prostate cancer cells promote ATM expression via recruitment of the androgen receptor to the ATM gene enhancer region [81]. Pancreatic cancer cells increase ATM expression by overexpressing the transcription factor CUX1 [82,83]. Melanoma cells increase ATM signaling by increasing MAGE-C2 levels, which associates with KAP1 and promotes its Ser824 phosphorylation [84].
Promoting ATM signaling and expression may facilitate cancer cells’ resistance to chemotherapy and radiation, metastasis, and tumor cell survival [79] (Figure 4). As radiation therapy and many chemotherapy agents attack cancer cells by causing DSBs, elevated ATM’s function and signaling provide tumors with significant advantages to adapt and survive these treatments. In fact, ATM-mediated activation of Akt can induce cancer cell survival in certain scenarios [82]. NF-KB activation by ATM additionally increases cancer cell survival, blocks apoptosis, and facilitates Epithelial-Mesenchymal Transition, as well as cancer cell migration and metastasis [85]. ATM also triggers the pentose phosphate pathway to reduce ROS levels. This metabolic pathway also provides cancer cells with essential intermediates for biosynthesis and proliferation [86–88]. Of note, increased ATM function has been found to be associated with elevated metastasis, invasion, and Epithelial-Mesenchymal Transition of breast cancer cells that overexpress HOXB9 or under-express PRSS11 [89–91]. Moreover, ATM is activated by hypoxia, and stabilizes the transcription factor HIF1α by direct phosphorylation or via the TRAF6/H2AX/HIF1α signaling axis, which could significantly increase cancer survival, invasion, and metastasis [58,63,79].
6. Conclusions
Numerous studies during the past three decades show that ATM is a central coordinator of the DNA-damage response and plays a vital role in maintaining genomic integrity and suppressing cancer at early stages. Indeed, ATM suppresses tumors by inducing apoptosis and cell cycle arrest. However, in some cancer cells that have already escaped those tumor-suppressing mechanisms, ATM signaling may be beneficial for cancer survival, resistance to chemotherapy and radiation, promoting Epithelial-Mesenchymal Transition, proliferation, invasion, and metastasis. Therefore, ATM-based anti-cancer therapy should be carefully selected and tailored based on the characteristics of the tumors to achieve significant and long-lasting therapeutic effects. Interestingly, beside its functions in DNA damage response and maintaining genomic integrity, ATM is also involved in oxidative stress sensing, regulating autophagy, mitophagy, pexophagy, and maintaining cellular homeostasis in response to ROS, starvation, and hypoxia. Together, these findings suggest a multi-faceted role of ATM in various cellular processes, which significantly expands its functions beyond guarding genome stability.
References
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; A Tagle, D.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753, doi:10.1126/science.7792600.
- Abraham, R.T. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair 2004, 3, 883–887, doi:10.1016/j.dnarep.2004.04.002.
- Paull, T.T. Mechanisms of ATM Activation. Rev. Biochem. 2015, 84, 711–738, doi:10.1146/annurev-biochem-060614-034335.
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008, doi:10.1016/j.dnarep.2009.04.006.
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196, doi:10.1101/gad.914401.
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–227, doi:10.1016/s0968-0004(00)01563-2.
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC Domains of PIKK Proteins Are Functionally Equivalent and Participate in the Tip60-dependent Activation of DNA-PKcs and ATM. Biol. Chem. 2006, 281, 15741–15746, doi:10.1074/jbc.m513172200.
- Fernandes, N.; Sun, Y.; Chen, S.; Paul, P.; Shaw, R.J.; Cantley, L.C.; Price, B.D. DNA Damage-induced Association of ATM with Its Target Proteins Requires a Protein Interaction Domain in the N Terminus of ATM. Biol. Chem. 2005, 280, 15158–15164, doi:10.1074/jbc.m412065200.
- Andrade, M.A.; Petosa, C.; I O’Donoghue, S.; Müller, C.W.; Bork, P. Comparison of ARM and HEAT protein repeats. Mol. Biol. 2001, 309, 1–18, doi:10.1006/jmbi.2001.4624.
- Piazza, I.; Rutkowska, A.; Ori, A.; Walczak, M.; Metz, J.; Pelechano, V.; Beck, M.; Haering, C.H. Association of condensin with chromosomes depends on DNA binding by its HEAT-repeat subunits. Struct. Mol. Biol. 2014, 21, 560–568, doi:10.1038/nsmb.2831.
- Rubinson, E.H.; Gowda, A.S.P.; Spratt, T.E.; Gold, B.; Eichman, B.F. An Unprecedented Nucleic Acid Capture Mechanism for Excision of DNA Damage. Nature 2010, 468, 406–411, doi:10.1038/nature09428.
- Dupré, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the Mre11–Rad50–Nbs1 complex. Struct. Mol. Biol. 2006, 13, 451–457, doi:10.1038/nsmb1090.
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611, doi:10.1038/nature03442.
- Kanu, N.; Behrens, A. ATMIN defines an NBS1-independent pathway of ATM signalling. EMBO J. 2007, 26, 2933–2941, doi:10.1038/sj.emboj.7601733.
- Bowen, C.; Ju, J.-H.; Lee, J.-H.; Paull, T.T.; Gelmann, E.P. Functional activation of ATM by the prostate cancer suppressor NKX3.1. Cell Rep. 2013, 4, 516–29, doi:10.1016/j.celrep.2013.06.039.
- Lavin, M.F. The Mre11 complex and ATM: A two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair 2004, 3, 1515–1520, doi:10.1016/j.dnarep.2004.07.001.
- Lavin, M.F.; Scott, S.; Gueven, N.; Kozlov, S.; Peng, C.; Chen, P. Functional consequences of sequence alterations in the ATM gene. DNA Repair 2004, 3, 1197–1205, doi:10.1016/j.dnarep.2004.03.011.
- Lee, J.H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554, doi:10.1126/science.1108297.
- Lee, J.H.; Paull, T.T. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science 2004, 304, 93–96, doi:10.1126/science.1091496.
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621, doi:10.1093/emboj/cdg541.
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Rev. Mol. Cell Biol. 2013, 14, 197–210, doi:10.1038/nrm3546.
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506, doi:10.1038/nature01368.
- Berkovich, E.; Monnat, R.J., Jr.; Kastan, M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Cell Biol. 2007, 9, 683–690, doi:10.1038/ncb1599.
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.-Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-Dependent and -Independent Dynamics of the Nuclear Phosphoproteome After DNA Damage. Signal. 2010, 3, rs3, doi:10.1126/scisignal.2001034.
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166, doi:10.1126/science.1140321.
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. J. Hum. Genet. 1986, 39, 573–83.
- Álvarez-Quilón, A.; Serrano-Benítez, A.; Lieberman, J.A.; Quintero, C.; Sánchez-Gutiérrez, D.; Escudero, L.M.; Cortés-Ledesma, F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Commun. 2014, 5, 3347, doi:10.1038/ncomms4347.
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776, doi:10.1038/sj.embor.7400210.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078, doi:10.1038/nature08467.
- Gatti, R.A.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.G.J.; Lange, K.; et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature 1988, 336, 577–580, doi:10.1038/336577a0.
- Taylor, A.M.; Byrd, P.J. Molecular pathology of ataxia telangiectasia. Clin. Pathol. 2005, 58, 1009–1015, doi:10.1136/jcp.2005.026062.
- Chun, H.H.; A Gatti, R. Ataxia–telangiectasia, an evolving phenotype. DNA Repair 2004, 3, 1187–1196, doi:10.1016/j.dnarep.2004.04.010.
- Renwick, A.; The Breast Cancer Susceptibility Collaboration (UK); Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Genet. 2006, 38, 873–875, doi:10.1038/ng1837.
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360, doi:10.1038/onc.2013.275.
- Wu, J.; Zhang, X.; Zhang, L.; Wu, C.-Y.; Rezaeian, A.H.; Chan, C.-H.; Li, J.-M.; Wang, J.; Gao, Y.; Han, F.; et al. Skp2 E3 Ligase Integrates ATM Activation and Homologous Recombination Repair by Ubiquitinating NBS1. Cell 2012, 46, 351–361, doi:10.1016/j.molcel.2012.02.018.
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. Cell Biol. 2009, 187, 977–990, doi:10.1083/jcb.200906064.
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446, doi:10.1016/j.cell.2008.12.041.
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell 2009, 136, 420–434, doi:10.1016/j.cell.2008.12.042.
- Kolas, N.K.; Chapman, R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science 2007, 318, 1637–1640, doi:10.1126/science.1150034.
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195, doi:10.1016/j.cell.2012.08.005.
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grøfte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 Suppress Spreading of Chromatin Ubiquitylation at Damaged Chromosomes. Cell 2012, 150, 697–709, doi:10.1016/j.cell.2012.06.039.
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent Initiation: A Mechanism for Triggering p53 Pulses in Response to DNA Damage. Cell 2008, 30, 277–289, doi:10.1016/j.molcel.2008.03.016.
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Cell Biol. 2006, 8, 870–876, doi:10.1038/ncb1446.
- Goodarzi, A.A.; Noon, A.T.; Jeggo, P.A. The impact of heterochromatin on DSB repair. Soc. Trans. 2009, 37, 569–576, doi:10.1042/bst0370569.
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.-Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Cell 2011, 41, 529–542, doi:10.1016/j.molcel.2011.02.015.
- Penicud, K.; Behrens, A. DMAP1 is an essential regulator of ATM activity and function. Oncogene 2014, 33, 525–531, doi:10.1038/onc.2012.597.
- Kim, Y.-C.; Gerlitz, G.; Furusawa, T.; Catez, F.; Nussenzweig, A.; Oh, K.-S.; Kraemer, K.H.; Shiloh, Y.; Bustin, M. Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Cell Biol. 2008, 11, 92–96, doi:10.1038/ncb1817.
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-Dependent Chromatin Changes Silence Transcription In cis to DNA Double-Strand Breaks. Cell 2010, 141, 970–981, doi:10.1016/j.cell.2010.04.038.
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; Van Lohuizen, M.; Ganesan, S. BMI1 Is Recruited to DNA Breaks and Contributes to DNA Damage-Induced H2A Ubiquitination and Repair. Cell. Biol. 2011, 31, 1972–1982, doi:10.1128/mcb.00981-10.
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. Cell Biol. 2010, 191, 45–60, doi:10.1083/jcb.201003034.
- Loizou, J.I.; Sancho, R.; Kanu, N.; Bolland, D.J.; Yang, F.; Rada, C.; Corcoran, A.E.; Behrens, A. ATMIN Is Required for Maintenance of Genomic Stability and Suppression of B Cell Lymphoma. Cancer Cell 2011, 19, 587–600, doi:10.1016/j.ccr.2011.03.022.
- Zhang, T.; Penicud, K.; Bruhn, C.; Loizou, J.I.; Kanu, N.; Wang, Z.-Q.; Behrens, A. Competition between NBS1 and ATMIN Controls ATM Signaling Pathway Choice. Cell Rep. 2012, 2, 1498–1504, doi:10.1016/j.celrep.2012.11.002.
- Kanu, N.; Penicud, K.; Hristova, M.; Wong, B.; Irvine, E.; Plattner, F.; Raivich, G.; Behrens, A. The ATM Cofactor ATMIN Protects against Oxidative Stress and Accumulation of DNA Damage in the Aging Brain. Biol. Chem. 2010, 285, 38534–38542, doi:10.1074/jbc.m110.145896.
- Burdak-Rothkamm, S.; Rothkamm, K.; Prise, K.M. ATM Acts Downstream of ATR in the DNA Damage Response Signaling of Bystander Cells. Cancer Res. 2008, 68, 7059–7065, doi:10.1158/0008-5472.can-08-0545.
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van IJcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58, doi:10.1038/nature14512.
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM Activation by Oxidative Stress. Science 2010, 330, 517–521, doi:10.1126/science.1192912.
- Alexander, A.; Cai, S.-L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.-L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Natl. Acad. Sci. USA 2010, 107, 4153–4158, doi:10.1073/pnas.0913860107.
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 Signaling under Hypoxic Conditions Is Controlled by ATM-Dependent Phosphorylation of HIF-1α. Cell 2010, 40, 509–520, doi:10.1016/j.molcel.2010.10.030.
- Hammond, E.M.; Kaufmann, M.R.; Giaccia, A.J. Oxygen sensing and the DNA-damage response. Opin. Cell Biol. 2007, 19, 680–684, doi:10.1016/j.ceb.2007.10.002.
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.-L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of Acute versus Chronic Hypoxia on DNA Damage Responses and Genomic Instability. Cancer Res. 2010, 70, 925–935, doi:10.1158/0008-5472.can-09-2715.
- Bencokova, Z.; Kaufmann, M.R.; Pires, I.M.; Lecane, P.S.; Giaccia, A.J.; Hammond, E.M. ATM Activation and Signaling under Hypoxic Conditions. Cell. Biol. 2008, 29, 526–537, doi:10.1128/mcb.01301-08.
- Olcina, M.M.; Foskolou, I.P.; Anbalagan, S.; Senra, J.M.; Pires, I.M.; Jiang, Y.; Ryan, A.J.; Hammond, E.M. Replication Stress and Chromatin Context Link ATM Activation to a Role in DNA Replication. Cell 2013, 52, 758–766, doi:10.1016/j.molcel.2013.10.019.
- Rezaeian, A.-H.; Li, C.-F.; Wu, C.-Y.; Zhang, X.; DeLacerda, J.; You, M.J.; Han, F.; Cai, Z.; Jeong, Y.S.; Jin, G.; et al. A hypoxia-responsive TRAF6–ATM–H2AX signalling axis promotes HIF1α activation, tumorigenesis and metastasis. Cell Biol. 2017, 19, 38–51, doi:10.1038/ncb3445.
- Stagni, V.; Ferri, A.; Cirotti, C.; Barilà, D. ATM Kinase-Dependent Regulation of Autophagy: A Key Player in Senescence? Cell Dev. Biol. 2021, 8, 599048, doi:10.3389/fcell.2020.599048.
- Stagni, V.; Cirotti, C.; Barilà, D. Ataxia-Telangiectasia Mutated Kinase in the Control of Oxidative Stress, Mitochondria, and Autophagy in Cancer: A Maestro with a Large Orchestra. Oncol. 2018, 8, 73, doi:10.3389/fonc.2018.00073.
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V. DNA Damage Response and Autophagy: A Meaningful Partnership. Genet. 2016, 7, 204, doi:10.3389/fgene.2016.00204.
- Alexander, A.; Walker, C.L. Differential localization of ATM is correlated with activation of distinct downstream signaling pathways. Cell Cycle 2010, 9, 3709–3710, doi:10.4161/cc.9.18.13253.
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673, doi:10.4161/auto.6.5.12509.
- Antonelli, M.; Strappazzon, F.; Arisi, I.; Brandi, R.; D’Onofrio, M.; Sambucci, M.; Manic, G.; Vitale, I.; Barilà, D.; Stagni, V. ATM kinase sustains breast cancer stem-like cells by promoting ATG4C expression and autophagy. Oncotarget 2017, 8, 21692–21709, doi:10.18632/oncotarget.15537.
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.L.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Cell Biol. 2015, 17, 1259–1269, doi:10.1038/ncb3230.
- Tripathi, D.N.; Zhang, J.; Jing, J.; Dere, R.; Walker, C.L. A new role for ATM in selective autophagy of peroxisomes (pexophagy). Autophagy 2016, 12, 711–712, doi:10.1080/15548627.2015.1123375.
- Shiloh, Y. ATM: Expanding roles as a chief guardian of genome stability. Cell Res. 2014, 329, 154–161, doi:10.1016/j.yexcr.2014.09.002.
- Lee, J.-H.; Paull, T.T. Mitochondria at the crossroads of ATM-mediated stress signaling and regulation of reactive oxygen species. Redox Biol. 2020, 32, 101511, doi:10.1016/j.redox.2020.101511.
- Liang, N.; He, Q.; Liu, X.; Sun, H. Multifaceted roles of ATM in autophagy: From nonselective autophagy to selective autophagy. Cell Biochem. Funct.2019, 37, 177–184, doi:10.1002/cbf.3385.
- Valentin-Vega, Y.A.; MacLean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.R.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500, doi:10.1182/blood-2011-08-373639.
- Guo, Q.; Wang, S.; Xu, H.; Li, X.; Guan, Y.; Yi, F.; Zhou, T.; Jiang, B.; Bai, N.; Ma, M.; et al. ATM—CHK 2‐Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111, doi:10.15252/embj.2019103111.
- Qi, Y.; Qiu, Q.; Gu, X.; Tian, Y.; Zhang, Y. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Rep.2016, 6, 24700, doi:10.1038/srep24700.
- Song, L.; Lin, C.; Wu, Z.; Gong, H.; Zeng, Y.; Wu, J.; Li, M.; Li, J. miR-18a Impairs DNA Damage Response through Downregulation of Ataxia Telangiectasia Mutated (ATM) Kinase. PLoS ONE 2011, 6, e25454, doi:10.1371/journal.pone.0025454.
- Rezaeian, A.-H.; Khanbabaei, H.; Calin, G.A. Therapeutic Potential of the miRNA-ATM Axis in the Management of Tumor Radioresistance. Cancer Res.2019, 80, 139–150, doi:10.1158/0008-5472.CAN-19-1807.
- Le Guezennec, X.; Bulavin, D.V. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 2010, 35, 109–114, doi:10.1016/j.tibs.2009.09.005.
- Mahajan, K.; Coppola, D.; Rawal, B.; Chen, Y.A.; Lawrence, H.R.; Engelman, R.W.; Lawrence, N.J.; Mahajan, N.P. Ack1-mediated Androgen Receptor Phosphorylation Modulates Radiation Resistance in Castration-resistant Prostate Cancer. Biol. Chem. 2012, 287, 22112–22122, doi:10.1074/jbc.m112.357384.
- Ripka, S.; Neesse, A.; Riedel, J.; Bug, E.; Aigner, A.; Poulsom, R.; Fulda, S.; Neoptolemos, J.; Greenhalf, W.; Barth, P.; et al. CUX1: Target of Akt signalling and mediator of resistance to apoptosis in pancreatic cancer. Gut 2010, 59, 1101–1110, doi:10.1136/gut.2009.189720.
- Vadnais, C.; Davoudi, S.; Afshin, M.; Harada, R.; Dudley, R.; Clermont, P.-L.; Drobetsky, E.; Nepveu, A. CUX1 transcription factor is required for optimal ATM/ATR-mediated responses to DNA damage. Nucleic Acids Res. 2012, 40, 4483–4495, doi:10.1093/nar/gks041.
- Bhatia, N.; Xiao, T.Z.; Rosenthal, K.A.; Siddiqui, I.A.; Thiyagarajan, S.; Smart, B.; Meng, Q.; Zuleger, C.L.; Mukhtar, H.; Kenney, S.C.; et al. MAGE-C2 Promotes Growth and Tumorigenicity of Melanoma Cells, Phosphorylation of KAP1, and DNA Damage Repair. Investig. Dermatol. 2013, 133, 759–767, doi:10.1038/jid.2012.355.
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261, doi:10.1158/0008-5472.can-11-3386.
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2010, 30, 546–555, doi:10.1038/emboj.2010.330.
- Krüger, A.; Grüning, N.-M.; Wamelink, M.M.; Kerick, M.; Kirpy, A.; Parkhomchuk, D.; Bluemlein, K.; Schweiger, M.-R.; Soldatov, A.; Lehrach, H.; et al. The Pentose Phosphate Pathway Is a Metabolic Redox Sensor and Regulates Transcription During the Antioxidant Response. Redox Signal. 2011, 15, 311–324, doi:10.1089/ars.2010.3797.
- Krüger, A.; Ralser, M. ATM Is a Redox Sensor Linking Genome Stability and Carbon Metabolism. Signal. 2011, 4, pe17, doi:10.1126/scisignal.2001959.
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.D.M.; Wijendran, V.; Shioda, T.; et al. HOXB9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Natl. Acad. Sci. USA 2010, 107, 1100–1105, doi:10.1073/pnas.0912710107.
- Wang, N.; Eckert, K.A.; Zomorrodi, A.R.; Xin, P.; Pan, W.; Shearer, D.A.; Weisz, J.; Maranus, C.D.; Clawson, G.A. Down-Regulation of HtrA1 Activates the Epithelial-Mesenchymal Transition and ATM DNA Damage Response Pathways. PLoS ONE 2012, 7, e39446, doi:10.1371/journal.pone.0039446.
- Chiba, N.; Comaills, V.; Shiotani, B.; Takahashi, F.; Shimada, T.; Tajima, K.; Winokur, D.; Hayashida, T.; Willers, H.; Brachtel, E.; et al. Homeobox B9 induces epithelial-to-mesenchymal transition-associated radioresistance by accelerating DNA damage responses. Natl. Acad. Sci. USA 2012, 109, 2760–2765, doi:10.1073/pnas.1s018867108.
This entry is adapted from the peer-reviewed paper 10.3390/genes12060845