Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
RNA-binding proteins (RBPs) are multi-faceted proteins in the regulation of RNA or its RNA splicing, localisation, stability, and translation.
- RNA binding protein
- splicing factor
- translation regulator
1. Introduction
RNA-binding proteins (RBPs) are critical RNA regulators responsible for modulating post-transcriptional events in the cell. RBPs can recognize and interact with binding motifs called RNA recognition motifs (RRM) and/or RNA structure to form ribonucleoprotein (RNP) complexes for the regulation of various RNA processes such as RNA stability, alternative pre-mRNA splicing, mRNA decay, translocation, post-translational nucleotide modifications, and RNA localization (Figure 1) [1][2].
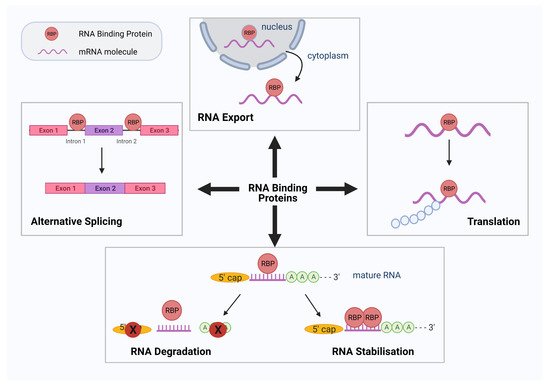
Figure 1. Schematic diagram summarizing the various roles of RNA binding proteins (RBPs). RBPs have numerous roles in RNA processing and translation. Four such functions of RBPs are demonstrated diagrammatically above: alternative splicing, RNA export, protein translation, RNA degradation, and stabilization. Figure created using Biorender.com.
2. RBPs in the Pathogenesis of Diabetes and Cardiovascular Disease
Diabetes mellitus (DM) is an increasingly prevalent global health burden [3]. DM is a lifelong disease, characterized by chronic hyperglycemia. DM is highly associated with an increased risk of debilitating secondary morbidities manifesting in macrovascular disease (atherosclerosis, ischemic stroke, coronary artery disease) and microvascular disease (diabetic retinopathy, neuropathy, and nephropathy) [4]. Close to 10% of worldwide diabetes diagnoses are categorized as Type 1. The remaining majority are diagnosed as Type 2 [5], where cells become increasingly resistant to insulin action [6], leading to impaired glucose homeostasis with cells unable to internalize circulating blood glucose. Chronic hyperglycemia causes systemic damage to the vasculature triggering multisystemic conditions such as cardiovascular disease (CVD). Additionally, due to the frequency of CVD occurrence in diabetes, it is often considered a CVD in itself. Currently, there is no curative therapy available for diabetes-associated CVD. With rising rates of diabetes, there lies a deepening need for knowledge into the mechanisms behind hyperglycemia-related cardiovascular damage. In vascular endothelial cells (ECs), hyperglycemia has been determined to contribute to a substantial change of gene expression. Transcriptomic analytical assays have uncovered a wide variety of candidate genes implicated in cellular functions such as angiogenesis, coagulation, vascular tone, adhesion, and more. This vascular EC gene expression is tightly controlled by transcriptional and post-transcriptional regulatory mechanisms, the latter including regulation of pre-mRNA to mRNA processing, transport, decay and protein translation [7]. Precise regulation of these complex post-transcriptional modifications in the RNA network is crucial for the normal function of vascular ECs and the endothelial system. In diabetes, a plethora of RBP-regulated RNA networks are involved in the dysfunction of the vascular endothelium [8]. In this section, we will review some of the most common RBPs dysregulated in the pathogenenesis of DM and CVD and their epigenetic effects.
For instance, RNA Binding Fox-1 Homolog 2 (RBFOX2), regulates alternative splicing and is upregulated in the diabetic heart, controlling splicing of genes involved in diabetic cardiomyopathy by binding to target RNA motifs associated with protein trafficking and cell apoptosis [9][10]. Additionally, Human Antigen R (HuR) also known with its alternative name ELAVL1, is a ubiquitously expressed RBP, which is upregulated and activated under high glucose and in diabetes [11]. HuR binds to specific domains known as AU-rich elements (ARE) in the 3′UTRs of target genes that play a role in inflammation and diabetic nephropathy [12][13][14]. Once it is activated, it translocates to the cytoplasm to bind its mRNA targets affecting their stability and translation [15]. Tristetraprolin (TTP) binds to 3′ UTR ARE region that results in mRNA destabilization and decay [16]. TTP is minimally expressed in healthy aortas but significantly heightened in affected macrophage foam cells as well as ECs of atherosclerotic lesions [17]. In another example, Quaking (QKI), an RBP member of the Signal Transduction and Activation of RNA (STAR) protein family and some of its isoforms—namely QKI5, QKI6, QKI7—have been associated with vascular development [16]. In our lab, we have previously shown that, compared to controls, there are reduced QKI5 levels in cardiac vessels of diabetic mice, therefore displaying the key status of QKI5 within the diabetic framework of vessel dysfunction. In addition, as we have also reported [18], QKI5 played a crucial role in differentiating ECs from induced pluripotent stem cells (iPSCs) via stabilization of VE-cadherin and Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) activation through Signal Transducer and Activator of Transcription 3 (STAT3) signaling. Furthermore, we showed QKI-7 to bind and promote mRNA target degradation such as of VE-cadherin, while the knockdown of QKI7 in a diabetic mouse model of hindlimb ischemia significantly restored reperfusion and blood flow in vivo [19].
RBPs are also implicated in the dysfunction of ECs under diabetic conditions in relation to their association with non-protein coding RNAs (ncRNAs), which include long noncoding RNAs (lncRNAs). The latter are, in fact, responsible for the preponderance of gene transcripts and act as positive or negative regulators based on their interactions with RBPs [20]. The importance of the RBP and lncRNAs system as a fundamental part of healthy cellular function through regulation of epigenetic machineries is largely acknowledged [21]. In recent years, data from numerous studies has showed a correlation between abnormal levels of lncRNAs and different diseases such as diabetes. For example, the levels of Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1) were significantly elevated both in vivo (in retinal ECs of a streptozotocin (STZ) diabetic rat model) and in vitro when human umbilical vein endothelial cells (HUVECs) were treated with high glucose. Short hairpin RNA (shRNA) knockdown of MALAT1 reduced vascular dysfunction and also decreased reactive oxygen species (ROS) levels in hyperglycemic ECs signifying its connection with diabetic retinopathy and EC dysfunction [22][23]. Similarly, under diabetic conditions, myocardial infarction associated transcript (MIAT) lncRNA is elevated, as data from studies of diabetic retinas and high-glucose treated ECs have shown. Furthermore, knockdown of MIAT reversed the dysfunction [24]. More such examples of lncRNA dysregulation in diabetic conditions exist as in the case of increased antisense noncoding RNA in the INK4 Locus (ANRIL) [18] promoting pathogenic angiogenesis [25] or in the case of Maternally Expressed Gene 3 (MEG3), which had reduced levels in the retinal ECs as shown in an STZ model of diabetic mice [26].
3. RBPs and Their Role in Neurodegenerative Disease
Even though the functional mechanisms of RBPs are still not fully elucidated, more recent evidence has indicated that RBPs are key players in the preservation and integrity of neurons. Any defects and alterations in the function of RBPs and in RNA metabolism arising from mutations can cause several neurodegenerative diseases that affect the central nervous system, such as frontotemporal lobar degeneration (FTD), amyotrophic lateral sclerosis (ALS), fragile X syndrome (FXS), or spinal muscular atrophy (SMA). Other diseases usually associated with aging and that can be affected by RBP dysregulation include Alzheimer’s disease (AD) and Parkinson’s disease (PD). The increasing aging global population has additionally resulted in an increase in the number of worldwide dementia cases, despite a relative decrease in developed countries [27]. In the case of ALS for instance, analytical investigations have revealed a strong genetic relationship between mutations of RBP-encoding genes like Ataxin 2 (ATXN2), Heterogeneous Nuclear Ribonucleoprotein A1 (hnRNPA1), Matrin 3 (MATR3) or TIA1 Cytotoxic Granule Associated RNA Binding Protein (TIA-1), and development and progression of the disease [28][29]. Likewise, in FTD, which shares many common characteristics with ALS [30][31], fragmentation of RBPs such as TAR DNA-binding protein 43 (TDP-43) in the cytoplasm, has been shown to advance the onset of the disease [32][33]. In the case of SMA, a serious motor neuron disease, small molecule drug analogs of RG-7916 (SMN-C2 or -C3) were found to selectively regulate alternative splicing of Survival of Motor Neuron 2 (SMN2) by binding to the gene’s pre-mRNA and increasing the affinity of the RBP Far Upstream Element Binding Protein 1 (FUBP1) to it [34]. Another neurological syndrome Paraneoplastic opsoclonus-myoclonus ataxia (POMA) is caused by autoantibody secretion against the RBP neuro-oncological ventral antigen 1 and 2 (Nova 1, Nova 2) [35], which are neuron-specific found in the nucleus and regulate RNA splicing [36]. In another example, myotonic dystrophy (MD) which commonly presents in patients as muscular degeneration, is also characterized by aberrant RNA splicing; CUG triplet repeat (CUGBP) has been specifically linked to MD through its interaction with myotonic dystrophy protein kinase (DMPK) mRNA [37].
These types of neurological diseases usually present with aggressive and irreversible characteristics that can prove devastating, and on many occasions even fatal, such as permanent neuron loss, which involves neural cells such as microglia and astrocytes. In neurodegenerative disease a great deal of attention has been concentrated on the different protein aggregates; however, it is of utmost importance to also focus on additional avenues that involve RNA and post transcriptional modifications as a pathogenic component of neurodegenerative disease [38][39]. In neurons, looking at the high incidence of RNA transport granules may explain why RBP dysfunction can initiate neuronal disease. The RNA granules creation and aggregate formation in a cell’s cytoplasm has been considered to be pathogenic in nature. During cellular stress, RBPs like those with low complexity domains (LCD), such as FUS RNA binding protein (FUS) or hnRNPA1 translocate from the nucleus, where they are usually present, to the cytoplasm and localize in granules [40][41]. Once there, they transiently form droplet organelles [42] with different functions based on their components [43]. Higher RBP concentrations can change these functions and lead to the polymerization of LCDs and the creation of amyloid-like fibers and insoluble aggregates [44]. Studies on these aggregates are giving rise to hypotheses that in neurons affected by dementias such as ALS or AD, disturbances such as mutations in RBPs play a role in impairing their regular physiological function. In the case of dementias such as AD, a study on RBP-containing stress granules showed their elevated accumulation in the brains of transgenic mice used as a model of tauopathy [45]. Furthermore, these granules have an interconnecting role with miRNAs, since the latter interact with RBP to regulate protein translation [46], adding an additional layer of complexity.
In general, mutations in the proteins associated with disease increase their propensity for higher aggregation, shifting the balance towards increased creation of more stable, less soluble and, thus, more persistent, stress granules, including secondary granules, which are usually associated with disease. Equally, approaches in neuroprotective therapeutics are directing their efforts against pathogenesis by reducing the creation of stress granules and restoring the balance [47].
This entry is adapted from the peer-reviewed paper 10.3390/biology10050366
References
- Liu-Yesucevitz, L.; Bassell, G.J.; Gitler, A.D.; Hart, A.C.; Klann, E.; Richter, J.D.; Warren, S.T.; Wolozin, B. Local RNA translation at the synapse and in disease. J. Neurosci. 2011, 31, 16086–16093.
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.L.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711–719.
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843.
- Leon, B.M.; Maddox, T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258.
- Mobasseri, M.; Shirmohammadi, M.; Amiri, T.; Vahed, N.; Hosseini Fard, H.; Ghojazadeh, M. Prevalence and incidence of type 1 diabetes in the world: A systematic review and meta-analysis. Health Promot. Perspect 2020, 10, 98–115.
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39.
- Whelan, J.T.; Hollis, S.E.; Cha, D.S.; Asch, A.S.; Lee, M.H. Post-transcriptional regulation of the Ras-ERK/MAPK signaling pathway. J. Cell. Physiol. 2012, 227, 1235–1241.
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345.
- Nutter, C.A.; Jaworski, E.A.; Verma, S.K.; Deshmukh, V.; Wang, Q.; Botvinnik, O.B.; Lozano, M.J.; Abass, I.J.; Ijaz, T.; Brasier, A.R.; et al. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep. 2016, 15, 2200–2213.
- Brosseau, J.P.; Lucier, J.F.; Nwilati, H.; Thibault, P.; Garneau, D.; Gendron, D.; Durand, M.; Couture, S.; Lapointe, E.; Prinos, P.; et al. Tumor microenvironment-associated modifications of alternative splicing. RNA 2014, 20, 189–201.
- Jeyabal, P.; Thandavarayan, R.A.; Joladarashi, D.; Suresh Babu, S.; Krishnamurthy, S.; Bhimaraj, A.; Youker, K.A.; Kishore, R.; Krishnamurthy, P. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem. Biophys. Res. Commun. 2016, 471, 423–429.
- Liu, S.; Huang, Z.; Tang, A.; Wu, X.; Aube, J.; Xu, L.; Xing, C.; Huang, Y. Inhibition of RNA-binding protein HuR reduces glomerulosclerosis in experimental nephritis. Clin. Sci. 2020, 134, 1433–1448.
- Shang, J.; Zhao, Z. Emerging role of HuR in inflammatory response in kidney diseases. Acta Biochim. Biophys. Sin. 2017, 49, 753–763.
- Shi, Q.; Lee, D.-Y.; Féliers, D.; Abboud, H.E.; Bhat, M.A.; Gorin, Y. Interplay between RNA-binding protein HuR and Nox4 as a novel therapeutic target in diabetic kidney disease. Mol. Metab. 2020, 36, 100968.
- Govindaraju, S.; Lee, B.S. Adaptive and maladaptive expression of the mRNA regulatory protein HuR. World J. Biol. Chem. 2013, 4, 111–118.
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-Wide Association Identifies Nine Common Variants Associated With Fasting Proinsulin Levels and Provides New Insights Into the Pathophysiology of Type 2 Diabetes. Diabetes 2011, 60, 2624–2634.
- Lai, W.S.; Carballo, E.; Thorn, J.M.; Kennington, E.A.; Blackshear, P.J. Interactions of CCCH Zinc Finger Proteins with mRNA: Binding of Tristetraprolin-Related Zinc Finger Proteins to Au-Rich Elements and Destabilization of mRNA. J. Biol. Chem. 2000, 275, 17827–17837.
- Congrains, A.; Kamide, K.; Ohishi, M.; Rakugi, H. ANRIL: Molecular Mechanisms and Implications in Human Health. Int. J. Mol. Sci. 2013, 14, 1278–1292.
- Yang, C.; Eleftheriadou, M.; Kelaini, S.; Morrison, T.; González, M.V.; Caines, R.; Edwards, N.; Yacoub, A.; Edgar, K.; Moez, A.; et al. Targeting QKI-7 in vivo restores endothelial cell function in diabetes. Nat. Commun. 2020, 11, 3812.
- He, R.-Z.; Luo, D.-X.; Mo, Y.-Y. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis. 2019, 6, 6–15.
- Yang, L.; Froberg, J.E.; Lee, J.T. Long noncoding RNAs: Fresh perspectives into the RNA world. Trends Biochem. Sci. 2014, 39, 35–43.
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014, 5, e1506.
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell. Mol. Med. 2015, 19, 1418–1425.
- Yan, B.; Yao, J.; Liu, J.-Y.; Li, X.-M.; Wang, X.-Q.; Li, Y.-J.; Tao, Z.-F.; Song, Y.-C.; Chen, Q.; Jiang, Q. lncRNA-MIAT Regulates Microvascular Dysfunction by Functioning as a Competing Endogenous RNA. Circ. Res. 2015, 116, 1143–1156.
- Zhang, B.; Wang, D.; Ji, T.-F.; Shi, L.; Yu, J.-L. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-κB signaling pathway in a rat model. Oncotarget 2017, 8, 17347–17359.
- Qiu, G.-Z.; Tian, W.; Fu, H.-T.; Li, C.-P.; Liu, B. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141.
- Langa, K.M.; Larson, E.B.; Crimmins, E.M.; Faul, J.D.; Levine, D.A.; Kabeto, M.U.; Weir, D.R. A Comparison of the Prevalence of Dementia in the United States in 2000 and 2012. JAMA Intern. Med. 2017, 177, 51–58.
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320.
- Xue, Y.C.; Ng, C.S.; Xiang, P.; Liu, H.; Zhang, K.; Mohamud, Y.; Luo, H. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 78.
- Mackenzie, I.R.A.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007.
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14.
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. Dis. Models Mech. 2017, 10, 509–518.
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666.
- Wang, J.; Schultz, P.G.; Johnson, K.A. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. USA 2018, 115, E4604–E4612.
- Buckanovich, R.J.; Posner, J.B.; Darnell, R.B. Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron 1993, 11, 657–672.
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215.
- Milne, C.A.; Hodgkin, J. ETR-1, a homologue of a protein linked to myotonic dystrophy, is essential for muscle development in Caenorhabditis elegans. Curr. Biol. 1999, 9, 1243–1246.
- Belzil, V.V.; Gendron, T.F.; Petrucelli, L. RNA-mediated toxicity in neurodegenerative disease. Mol. Cell. Neurosci. 2013, 56, 406–419.
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438.
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473.
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372.
- Courchaine, E.M.; Lu, A.; Neugebauer, K.M. Droplet organelles? EMBO J. 2016, 35, 1603–1612.
- Smith, J.; Calidas, D.; Schmidt, H.; Lu, T.; Rasoloson, D.; Seydoux, G. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. Elife 2016, 5, e21337.
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 2015, 163, 829–839.
- Vanderweyde, T.; Yu, H.; Varnum, M.; Liu-Yesucevitz, L.; Citro, A.; Ikezu, T.; Duff, K.; Wolozin, B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J. Neurosci. 2012, 32, 8270–8283.
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149.
- Wolozin, B.; Apicco, D. RNA binding proteins and the genesis of neurodegenerative diseases. Adv. Exp. Med. Biol. 2015, 822, 11–15.
This entry is offline, you can click here to edit this entry!