Ferritin is one of the most frequently requested laboratory tests in primary and secondary care, and levels often deviate from reference ranges. Serving as an indirect marker for total body iron stores, low ferritin is highly specific for iron deficiency. Hyperferritinemia is, however, a non-specific finding, which is frequently overlooked in general practice. In routine medical practice, only 10% of cases are related to an iron overload, whilst the rest is seen as a result of acute phase reactions and reactive increases in ferritin due to underlying conditions. Differentiation of the presence or absence of an associated iron overload upon hyperferritinemia is essential, although often proves to be complex.
- ferritin
- iron
- inflammation
- hemochromatosis
1. Introduction
Ferritin is one of the most commonly requested laboratory tests in general and secondary care, and levels deviating from reference ranges are a frequent finding [1]. Ascribed to its proportionality to total body iron stores, ferritin function is an indirect marker of iron status [2]. When concurrent inflammation is absent, ferritin has proven to be a highly specific and sensitive parameter for the diagnosis of iron deficiency [3][4]. High ferritin, hyperferritinemia, may indicate increased iron stores, but is more commonly seen upon acute phase reactions and as a result of ferritin being released from damaged cells such as hepatocytes in liver disease [5]. It may also be the result of increased synthesis and/or increased cellular secretion of ferritin upon various stimuli such as cytokines, oxidants, hypoxia, oncogenes, and growth factors [6].
Ferritin reference ranges may vary according to the analytical assay being used, although upper cut off is typically set to 200 μg/L in women and 300 μg/L in men [1][7]. In a prospective Danish population-based study, ferritin proved to be a strong predictor of premature death in the general population. Subjects with a baseline ferritin ≥200 μg/L were found to have increased risk of cause-specific mortality due to cancer, endocrinological disease, and cardiovascular disease, as well as increased total mortality compared to those with levels <200 μg/L. The study furthermore found a stepwise increase of this risk upon stepwise increases in ferritin, with the highest cumulative risk seen upon levels ≥600 μg/L [8].
Clinical interpretation of hyperferritinemia often proves to be complex, and ferritin >1000 μg/L is regarded as a non-specific marker of pathology. General practitioners seem somewhat unfamiliar with the appropriate management of hyperferritinemia, as >50% of primary care patients presenting with ferritin levels of such magnitude, and without any obvious clinical reason, are not referred to secondary care nor offered any further investigation [9]. Based on the wide etiological spectrum, hyperferritinemia should prompt for further investigation through clinical examination and additional laboratory tests when the cause remains unknown [1][7].
2. Ferritin and Iron Homeostasis
Ferritin is mostly found as a cytosolic protein, where it plays an important role in the storage of intracellular iron, sequestering up to 4500 Fe3+ atoms per molecule. It is a 24-subunit molecule composed of two structurally distinct subunits, the light-chain (molecular weight 19 kilodalton) and the heavy-chain (molecular weight 21 kilodalton) [10]. Ferritin levels are upregulated and matched to intracellular iron levels through the activated translation of heavy- and light-chain mRNA upon high intracellular iron levels. The opposite is true upon low intracellular iron levels, resulting in prevented translation through specific sequences of heavy- and light-chain mRNA that block the recruitment of ribosomal subunits, as illustrated in Figure 1 [11].
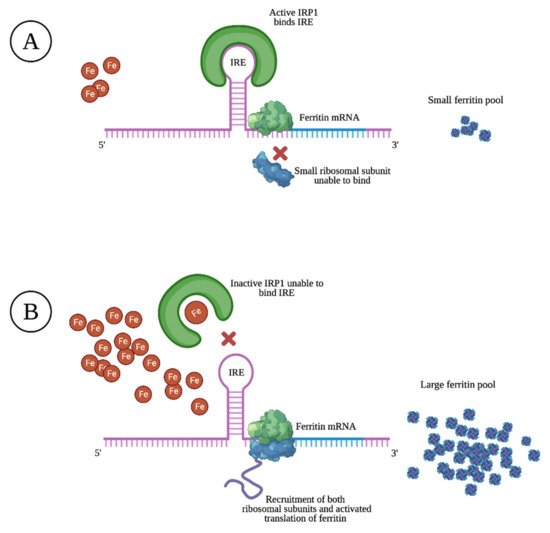
Within cells, limited amounts of free iron are found in a labile pool, which is biologically active in metabolism, although toxic if present in excess. By capturing and buffering iron, ferritin plays a key role in maintaining iron homeostasis [12]. Through its ferroxidase activity, heavy-chain ferritin subunits transform ferrous iron (Fe2+) into the less toxic ferric state (Fe3+). Homozygous murine knockouts of heavy-chain ferritin were found to be embryonically lethal, illustrating its importance in cellular defense, the detoxification of iron, and in maintaining iron bioavailability [13].
Through mechanisms that are not fully understood, small quantities of ferritin emerge into the serum. Controversial results regarding the subunit composition and iron content of ferritin found in serum are reported [14][15][16]. It is, however, assumed to be relatively iron-poor and almost entirely made up of light-chain subunits [6][17]. In the normal state, 50–80% of serum ferritin is glycosylated as a result of release from macrophages of the reticuloendothelial system (RES), and to some extent hepatocytes [1][18][19]. In certain hereditary hyperferritinemic states, glycosylation is almost 100% [20]. This supports a regulated release mechanism, whilst a higher percentage of non-glycosylated ferritin upon liver necrosis suggests hyperferritinemia to be the result of passive release, mainly due to cellular damage [1].
A schematic overview of the human iron metabolism is shown in Figure 2. The uptake of heme and non-heme iron takes place through the enterocytes of the proximal duodenum. Once inside the enterocyte, iron has two possible pathways—one portion remains intracellularly for use or for storage, while the rest is transported across the basolateral membrane through the iron exporter ferroportin, upon which it subsequently binds to transferrin. Effective efflux of iron through the basolateral membrane requires iron to be oxidized. This is mainly facilitated by the ferroxidase activity of plasma ceruloplasmin, which is synthesized in the liver, and its membrane-bound intestinal homolog hephaestin [21].
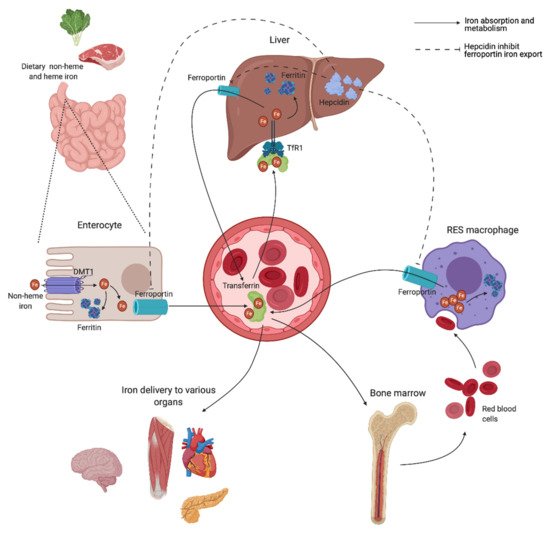
Transferrin is capable of sequestering only two iron atoms, and is also predominantly synthesized in the liver [22]. It is the major serum iron-binding protein, keeping iron biologically accessible in an aqueous environment for delivery to cells through the transferrin receptor 1 (TfR1), allowing Fe2+ atoms to be internalized through receptor-mediated endocytosis. Almost all tissue and cells express TfR1. Cells with increased iron demand have a particularly high expression, including rapidly dividing cells such as activated lymphocytes, as well as erythroblasts, which depend on iron delivery for hemoglobin production in erythropoiesis [21][23].
Ferroportin is also expressed in macrophages of the RES, involved in the recycling of iron from the hemoglobin of old red blood cells (RBCs) [24]. Irrespective of levels, iron is eliminated at a basal rate through the desquamation of skin and intestinal epithelium, and through blood loss in fertile women [25]. Both intracellular ferritin and the intestinal absorption of iron are important regulators of iron homeostasis, due to a lack of active physiological iron excretion mechanisms. Systemic regulation of metabolism and the modulation of availability to meet iron needs are predominantly mediated through the peptide hormone hepcidin and the hepcidin–ferroportin axis (Figure 2).
Hepcidin is mainly synthesized by hepatocytes, and facilitates synchronized iron metabolism between various organs, protecting the body against iron overload [26]. It is considered a negative regulator of serum iron levels as it mediates ubiquitin-mediated degradation of ferroportin, which shuts off export of iron to plasma. Iron is consequently retained in the intestinal epithelium, while iron recovery from senescent RBCs is interrupted by inhibited release from RES macrophages [21]. Hepcidin also facilitates a reduction in iron uptake by enterocytes through inhibited transcription and increased degradation of the intestinal iron transporter divalent metal transporter 1 (DMT1) [27][28]. Dysregulation of the hepcidin–ferroportin axis, promoting uncontrolled intestinal absorption and uninhibited cellular export of iron, is a characteristic feature in various iron-loading conditions exhibiting high serum iron levels [29][30], and is later discussed.
Finally, whether hyperferritinemia caused by disease processes has a causal role or a role in cellular protection is not fully established yet. Apart from ferritin’s iron-storing properties, the biological purpose of it remains partly unknown. Nevertheless, increasing evidence of ferritin subsuming additional physiological roles is emerging, and it is now suggested as molecule contributing to inflammation, iron delivery and angiogenesis, as well as cell signaling, proliferation and differentiation [18][21]. Increased understanding of these mechanisms might grant additional insight into the pathophysiology of iron overload, cancer, and inflammation, which may contribute to the development of novel therapeutic targets.
3. Etiology
Underlying conditions upon hyperferritinemia, with and without an associated iron overload, are summed up in Table 1. Transferrin saturation is a useful parameter for the distinction of the presence or absence of an iron overload upon hyperferritinemia. It is a calculated value reflecting the proportion of iron-binding sites on transferrin that are occupied [7]. Whilst a normal transferrin saturation usually excludes pathologically increased iron absorption, it does not necessarily exclude the presence of an iron overload [1][31][32][33]. Increases in transferrin saturation are not always equivalent to an iron overload either, and the interpretation of this parameter therefore requires careful considerations [29].
| Hyperferritinemia without iron overload | Common causes |
| Cellular damage | |
| Metabolic syndrome and obesity | |
| Insulin resistance/diabetes mellitus | |
| Excessive alcohol consumption | |
| Inflammatory and infectious conditions (septic shock, COVID-19) | |
| Malignancy (solid and hematological) | |
| Rare causes | |
| Benign hyperferritinemia/HHCS | |
| Immune-mediated syndromes (primary and secondary HLH, adult-onset Still’s disease) Gaucher disease |
|
| Hyperferritinemia with or without iron overload | Common causes |
| Chronic liver disease (cirrhosis, alcoholic liver disease, NAFLD, viral hepatitis, porphyria cutanea tarda) | |
| Hyperferritinemia with iron overload | Common causes |
| HFE hemochromatosis | |
| Dysmetabolic iron overloading syndrome | |
| Iron-loading anemias (congenital or acquired) | |
| Iatrogenic iron overload (RBC transfusion, parenteral iron administration) | |
| African iron overload | |
| Rare causes | |
| Non-HFE hereditary hemochromatosis Ferroportin disease |
|
| Aceruloplasminemia/hypoceruloplasminemia Atransferrinemia/hypotransferrinemia |
This differentiation of hyperferritinemia is essential, as the management, treatment, and prognosis greatly differ for the two entities. Estimates show that only 10% of clinical cases of hyperferritinemia in routine medical practice are associated with an iron overload. For the rest, one of the following underlying causes attributing to a reactive increase are usually identified: inflammation, metabolic syndrome, chronic alcohol consumption, cellular damage, and malignancy [34][35][36].
3.1. Hyperferritinemia without Iron Overload
3.2. Hyperferritinemia with Iron Overload
Iron overloading diseases are classified as (1) primary when caused by an inherited defect in the regulation of iron balance and (2) secondary when acquired as a result of underlying congenital or acquired conditions [70]. During iron-loading conditions where the iron binding capacity of transferrin is exceeded and a high transferrin saturation is observed, non-transferrin bound iron (NTBI) enters circulation. NTBI predominantly enters hepatocytes, but also the parenchymal cells of the heart, pancreas, thyroid, and central nervous system. This diffuse distribution of iron is associated with organ dysfunction through cell death and complications such as fibrosis, atherosclerosis, and carcinogenesis [23][30][71].
3.2.1. Primary Iron Overload
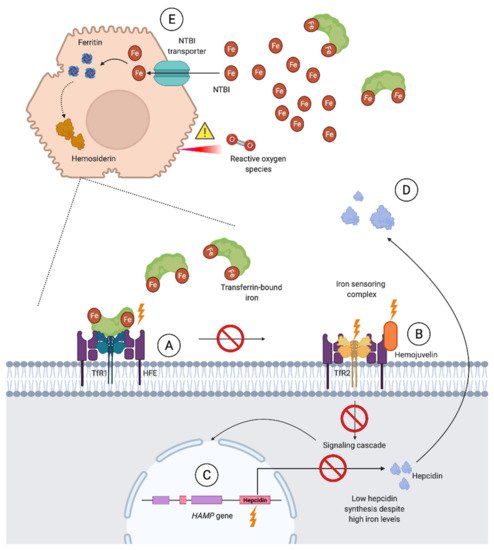
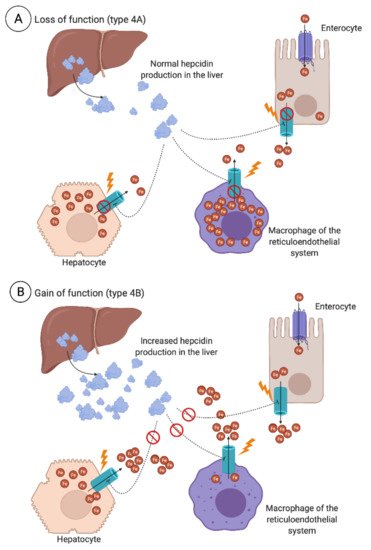
3.2.2. Secondary Iron Overload
Secondary iron overload occurs in individuals who absorb or store excessive amounts of iron as a result of underlying diseases other than those previously mentioned or due to iatrogenic iron overload through frequent RBC transfusions or parenteral iron administration. Owing to the liver’s major role in iron homeostasis, hyperferritinemia in liver diseases may also be associated with an iron overload. Reduced liver function in chronic liver diseases is associated with impaired hepcidin and transferrin synthesis, hepatic iron overload (HIO), as well as increased levels of circulating NTBI [50][91]. Consequently, iron overload has been proposed as a cofactor in these conditions, but its exact role remains unclear [92]. While HH may cause severe iron overload if left untreated, secondary iron overload due to chronic liver diseases usually remains minimal to modest [93].
HIO is present in up to 50% of patients with alcoholic liver disease [50]. Cirrhosis due to chronic hepatitis C contributes to hepatic iron accumulation [50], and HFE variants such as C282Y heterozygosity have shown to accelerate hepatic fibrosis and HIO in these patients [94]. Porphyria cutanea tarda should be considered when hyperferritinemia is seen with a photosensitive rash and liver dysfunction. This is an acquired liver disease in 80% of clinical cases, and exogenous factors (e.g., alcohol, smoking, and hepatitis C) are a prerequisite for inducing HIO in these patients [1][48][95].
One third of patients with NAFLD and metabolic syndrome have increased body iron stores, known as dysmetabolic iron overload syndrome—a much more common condition than clinically recognized by physicians. These patients most often exhibit a normal transferrin saturation and have been found to have increased hepcidin concentrations with a mixed pattern of iron retention in both hepatocytes and macrophages [96]. Studies suggest this to be the result of hepcidin-resistance and a compensatory mechanism to prevent and counteract iron accumulation [96][97][98].
Although iron deficiency anemia affects nearly all chronic kidney disease and long term hemodialysis patients as the disease progress [99], iatrogenic iron overload attributed to RBCs transfusions due to hypoproliferative erythroid marrow and intravenous iron to ensure sufficient available iron during therapy is also recognized as a complication in some of these patients. Mild to severe HIO measured by MRI was observed in 84% of hemodialysis patients treated with erythropoietin and regular intravenous iron supplementation, in keeping with guidelines, although transferrin saturation was normal for all [100]. Hyperferritinemia seen in chronic kidney disease patients on hemodialysis is also partly a result of systemic inflammation, with ferritin levels correlating positively with the severity of it [101].
Prolonged parenteral administration of iron or the transfusion of RBCs in patients with chronic anemia such as thalassemias or dyserythropoietic, aplastic, sideroblastic, and hemolytic anemias will also most often result in iron overload. Tissue deposition becomes significant when more than 40 units of RBCs are transfused [23]. Iron is deposited in the macrophages of the RES prior to the iron-loading of the liver and heart parenchyma, ultimately leading to progressive heart and liver failure if left untreated [91].
Even in the absence of transfusions, a variety of blood cell disorders such as myelodysplastic syndrome, thalassemias, and other iron-loading anemias are associated with increased iron absorption. Hepcidin is down-regulated by signaling molecules associated with anemia and hypoxia upon these conditions [30]. Ineffective erythropoiesis, therefore, leads to low hepcidin levels, and, subsequently, increased intestinal iron absorption. The hormone erythroferrone, secreted by erythroid precursors, suppresses hepcidin and increases the amount of iron available for hemoglobin synthesis [102]. Loss of this hormone in thalassemic knock out mice led to the full restoration of hepcidin mRNA expression and a significant reduction in the iron content of the liver and spleen [103], making erythroferrone a potential therapeutic target upon secondary iron overload in anemias and other blood cell disorders.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10092008
References
- Cullis, J.O.; Fitzsimons, E.J.; Griffiths, W.J.; Tsochatzis, E.; Thomas, D.W.; Haematology, T.B.S.F. Investigation and management of a raised serum ferritin. Br. J. Haematol. 2018, 181, 331–340.
- Jacobs, A.; Miller, F.; Worwood, M.; Beamish, M.R.; Wardrop, C.A. Ferritin in the serum of normal subjects and patients with iron deficiency and iron overload. Br. Med. J. 1972, 4, 206–208.
- Knovich, M.A.; Storey, J.A.; Coffman, L.G.; Torti, S.V.; Torti, F.M. Ferritin for the clinician. Blood Rev. 2009, 23, 95–104.
- Guyatt, G.H.; Oxman, A.D.; Ali, M.; Willan, A.; McIlroy, W.; Patterson, C. Laboratory diagnosis of iron-deficiency anemia: An overview. J. Gen. Intern. Med. 1992, 7, 145–153.
- Beaton, M.D.; Adams, P.C. Treatment of hyperferritinemia. Ann. Hepatol. 2012, 11, 294–300.
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516.
- Adams, P.C.; Barton, J.C. A diagnostic approach to hyperferritinemia with a non-elevated transferrin saturation. J. Hepatol. 2011, 55, 453–458.
- Ellervik, C.; Marott, J.L.; Tybjaerg-Hansen, A.; Schnohr, P.; Nordestgaard, B.G. Total and cause-specific mortality by moderately and markedly increased ferritin concentrations: General population study and metaanalysis. Clin. Chem. 2014, 60, 1419–1428.
- Ogilvie, C.; Fitzsimons, K.; Fitzsimons, E.J. Serum ferritin values in primary care: Are high values overlooked? J. Clin. Pathol. 2010, 63, 1124–1126.
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203.
- Hentze, M.; Caughman, S.; Rouault, T.; Barriocanal, J.; Dancis, A.; Harford, J.; Klausner, R. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 1987, 238, 1570–1573.
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559–1566.
- Ferreira, C.; Bucchini, D.; Martin, M.-E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024.
- Arosio, P.; Yokota, M.; Drysdale, J.W. Characterization of serum ferritin in iron overload: Possible identity to natural apoferritin. Br. J. Haematol. 1977, 36, 199–207.
- ten Kate, J.; Wolthuis, A.; Westerhuis, B.; van Deursen, C. The iron content of serum ferritin: Physiological importance and diagnostic value. Eur. J. Clin. Chem. Clin. Biochem. 1997, 35, 53–56.
- Cohen, L.A.; Gutiérrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.-L.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584.
- Santambrogio, P.; Cozzi, A.; Levi, S.; Arosio, P. Human serum ferritin G-peptide is recognized by anti-L ferritin subunit antibodies and concanavalin-A. Br. J. Haematol. 1987, 65, 235–237.
- Wang, W.; Knovich, M.A.; Coffman, L.G.; Torti, F.M.; Torti, S.V. Serum ferritin: Past, present and future. Biochim. Biophys. Acta 2010, 1800, 760–769.
- Ghosh, S.; Hevi, S.; Chuck, S.L. Regulated secretion of glycosylated human ferritin from hepatocytes. Blood 2004, 103, 2369–2376.
- Kannengiesser, C.; Jouanolle, A.-M.; Hetet, G.; Mosser, A.; Muzeau, F.; Henry, D.; Bardou-Jacquet, E.; Mornet, M.; Brissot, P.; Deugnier, Y.; et al. A new missense mutation in the L ferritin coding sequence associated with elevated levels of glycosylated ferritin in serum and absence of iron overload. Haematologica 2009, 94, 335–339.
- Andrews, N.C. Forging a field: The golden age of iron biology. Blood 2008, 112, 219–230.
- Anderson, E.R.; Shah, Y.M. Iron homeostasis in the liver. Compr. Physiol. 2013, 3, 315–330.
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15.
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200.
- Wallace, D.F. The regulation of iron absorption and homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62.
- Harrison-Findik, D.D. Role of alcohol in the regulation of iron metabolism. World J. Gastroenterol. 2007, 13, 4925–4930.
- Mena, N.P.; Esparza, A.; Tapia, V.; Valdes, P.; Nunez, M.T. Hepcidin inhibits apical iron uptake in intestinal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, 192–198.
- Brasse–Lagnel, C.; Karim, Z.; Letteron, P.; Bekri, S.; Bado, A.; Beaumont, C. Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterology 2011, 140, 1261–1271 e1.
- Adams, P.C.; Barton, J.C. Haemochromatosis. Lancet 2007, 370, 1855–1860.
- Fleming, R.E.; Ponka, P. Iron overload in human disease. N. Engl. J. Med. 2012, 366, 348–359.
- Bacon, B.R.; Adams, P.C.; Kowdley, K.V.; Powell, L.W.; Tavill, A.S. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 328–343.
- Adams, P.C.; Reboussin, D.M.; Barton, J.C.; McLaren, C.E.; Eckfeldt, J.H.; McLaren, G.D.; Dawkins, F.W.; Acton, R.T.; Harris, E.L.; Gordeuk, V.R.; et al. Hemochromatosis and iron-overload screening in a racially diverse population. N. Engl. J. Med. 2005, 352, 1769–1778.
- Koperdanova, M.; Cullis, J.O. Interpreting raised serum ferritin levels. BMJ 2015, 351, 3692.
- Hearnshaw, S.; Thompson, N.P.; McGill, A. The epidemiology of hyperferritinaemia. World J. Gastroenterol. 2006, 12, 5866–5869.
- Olynyk, J.K.; Cullen, D.J.; Aquilia, S.; Rossi, E.; Summerville, L.; Powell, L.W. A population-based study of the clinical expression of the hemochromatosis gene. N. Engl. J. Med. 1999, 341, 718–724.
- European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J. Hepatol. 2010, 53, 3–22.
- Tran, T.N.; Eubanks, S.K.; Schaffer, K.J.; Zhou, C.Y.; Linder, M.C. Secretion of ferritin by rat hepatoma cells and its regulation by inflammatory cytokines and iron. Blood 1997, 90, 4979–4986.
- Roy, C.N.; Andrews, N.C. Anemia of inflammation: The hepcidin link. Curr. Opin. Hematol. 2005, 12, 107–111.
- Ganz, T. Anemia of Inflammation. N. Engl. J. Med. 2019, 381, 1148–1157.
- Kernan, K.F.; Carcillo, J.A. Hyperferritinemia and inflammation. Int. Immunol. 2017, 29, 401–409.
- Rosario, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.P.; Shoenfeld, Y. The hyperferritinemic syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185.
- Lin, T.F.; Ferlic-Stark, L.L.; Allen, C.E.; Kozinetz, C.A.; McClain, K.L. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr. Blood Cancer 2011, 56, 154–155.
- Allen, C.E.; Yu, X.; Kozinetz, C.A.; McClain, K.L. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2008, 50, 1227–1235.
- Schram, A.M.; Campigotto, F.; Mullally, A.; Fogerty, A.; Massarotti, E.; Neuberg, D.; Berliner, N. Marked hyperferritinemia does not predict for HLH in the adult population. Blood 2015, 125, 1548–1552.
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034.
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062.
- Cheng, L.; Li, H.; Li, L.; Liu, C.; Yan, S.; Chen, H.; Li, Y. Ferritin in the coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. J. Clin. Lab. Anal. 2020, 34, e23618.
- Lorcerie, B.; Audia, S.; Samson, M.; Millière, A.; Falvo, N.; Leguy-Seguin, V.; Berthier, S.; Bonnotte, B. Diagnosis of hyperferritinemia in routine clinical practice. Press Med. 2017, 46, e329–e338.
- Kell, D.B.; Pretorius, E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 2014, 6, 748–773.
- Milic, S.; Mikolasevic, I.; Orlic, L.; Devcic, E.; Starcevic-Cizmarevic, N.; Stimac, D.; Kapovic, M.; Ristic, S. The Role of Iron and Iron Overload in Chronic Liver Disease. Med. Sci. Monit. 2016, 22, 2144–2151.
- Moirand, R.; Lescoat, G.; Delamaire, D.; Lauvin, L.; Campion, J.P.; Deugnier, Y.; Brissot, P. Increase in glycosylated and nonglycosylated serum ferritin in chronic alcoholism and their evolution during alcohol withdrawal. Alcohol Clin. Exp. Res. 1991, 15, 963–969.
- Moirand, R.; Kerdavid, F.; Loréal, O.; Hubert, N.; Leroyer, P.; Brissot, P.; Lescoat, G. Regulation of ferritin expression by alcohol in a human hepatoblastoma cell line and in rat hepatocyte cultures. J. Hepatol. 1995, 23, 431–439.
- Harrison-Findik, D.D.; Klein, E.; Crist, C.; Evans, J.; Timchenko, N.; Gollan, J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 2007, 46, 1979–1985.
- Ioannou, G.N.; Weiss, N.S.; Kowdley, K.V. Relationship between transferrin-iron saturation, alcohol consumption, and the incidence of cirrhosis and liver cancer. Clin. Gastroenterol. Hepatol. 2007, 5, 624–629.
- Ioannou, G.N.; Dominitz, J.A.; Weiss, N.S.; Heagerty, P.J.; Kowdley, K.V. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology 2004, 126, 1293–1301.
- Whitfield, J.B.; Zhu, G.; Heath, A.C.; Powell, L.W.; Martin, N.G. Effects of alcohol consumption on indices of iron stores and of iron stores on alcohol intake markers. Alcohol Clin. Exp. Res. 2001, 25, 1037–1045.
- Conte, D.; Corsetti, M.; Colli, A.; Bardella, M.T.; Cocciolo, M.; Fraquelli, A.M. Iron-related indexes in chronic alcoholics. Effect of alcohol withdrawal. Ital. J. Gastroenterol. Hepatol. 1998, 30, 534–538.
- Brudevold, R.; Hole, T.; Hammerstrom, J. Hyperferritinemia is associated with insulin resistance and fatty liver in patients without iron overload. PLoS ONE 2008, 3, e3547.
- Jehn, M.; Clark, J.M.; Guallar, E. Serum ferritin and risk of the metabolic syndrome in U.S. adults. Diabetes Care 2004, 27, 2422–2428.
- Haap, M.; Fritsche, A.; Mensing, H.J.; Haring, H.U.; Stumvoll, M. Association of high serum ferritin concentration with glucose intolerance and insulin resistance in healthy people. Ann. Intern. Med. 2003, 139, 869–871.
- Bozzini, C.; Girelli, D.; Olivieri, O.; Martinelli, N.; Bassi, A.; De Matteis, G.; Tenuti, I.; Lotto, V.; Friso, S.; Pizzolo, F.; et al. Prevalence of body iron excess in the metabolic syndrome. Diabetes Care 2005, 28, 2061–2063.
- Trombini, P.; Piperno, A. Ferritin, metabolic syndrome and NAFLD: Elective attractions and dangerous liaisons. J. Hepatol. 2007, 46, 549–552.
- Tuomainen, T.-P.; Nyyssönen, K.; Salonen, R.; Tervahauta, A.; Korpela, H.; Lakka, T.; A Kaplan, G.; Salonen, J.T. Body iron stores are associated with serum insulin and blood glucose concentrations. Population study in 1,013 eastern Finnish men. Diabetes Care 1997, 20, 426–428.
- Beaton, M.D.; Chakrabarti, S.; Adams, P.C. Inflammation is not the cause of an elevated serum ferritin in non-alcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 353–356.
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Halpern, Z.; Oren, R. NAFLD and hyperinsulinemia are major determinants of serum ferritin levels. J. Hepatol. 2007, 46, 700–707.
- Cadenas, B.; Fita-Torró, J.; Bermúdez-Cortés, M.; Hernandez-Rodriguez, I.; Fuster, J.L.; Llinares, M.E.; Galera, A.M.; Romero, J.L.; Pérez-Montero, S.; Tornador, C.; et al. L-Ferritin: One gene, five diseases; from hereditary hyperferritinemia to hypoferritinemia—Report of new cases. Pharmaceuticals 2019, 12, 17.
- Thurlow, V.; Vadher, B.; Bomford, A.; Delord, C.; Kannengiesser, C.; Beaumont, C.; Grandchamp, B. Two novel mutations in the L ferritin coding sequence associated with benign hyperferritinaemia unmasked by glycosylated ferritin assay. Ann. Clin. Biochem. 2012, 49, 302–305.
- Mistry, P.K.; Sadan, S.; Yang, R.; Yee, J.; Yang, M. Consequences of diagnostic delays in type 1 Gaucher disease: The need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am. J. Hematol. 2007, 82, 697–701.
- Nagral, A. Gaucher disease. J. Clin. Exp. Hepatol. 2014, 4, 37–50.
- Piperno, A. Classification and diagnosis of iron overload. Haematologica 1998, 83, 447–455.
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995.
- Ogilvie, C.; Gaffney, D.; Murray, H.; Kerry, A.; Haig, C.; Spooner, R.; Fitzsimons, E.J. Improved detection of hereditary haemochromatosis. J. Clin. Pathol. 2015, 68, 218–221.
- Wood, M.J.; Skoien, R.; Powell, L.W. The global burden of iron overload. Hepatol. Int. 2009, 3, 434–444.
- Grosse, S.D.; Gurrin, L.C.; Bertalli, N.A.; Allen, K.J. Clinical penetrance in hereditary hemochromatosis: Estimates of the cumulative incidence of severe liver disease among HFE C282Y homozygotes. Genet. Med. 2018, 20, 383–389.
- Adams, P.C. Nonexpressing homozygotes for C282Y hemochromatosis: Minority or majority of cases? Mol. Genet. Metab. 2000, 71, 81–86.
- Allen, K.J.; Gurrin, L.C.; Constantine, C.C.; Osborne, N.J.; Delatycki, M.B.; Nicoll, A.J.; McLaren, C.E.; Bahlo, M.; Nisselle, A.E.; Vulpe, C.D.; et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N. Engl. J. Med. 2008, 358, 221–230.
- Walsh, A.; Dixon, J.L.; Ramm, G.A.; Hewett, D.G.; Lincoln, D.J.; Anderson, G.J.; Subramaniam, V.N.; Dodemaide, J.; Cavanaugh, J.A.; Bassett, M.L.; et al. The clinical relevance of compound heterozygosity for the C282Y and H63D substitutions in hemochromatosis. Clin. Gastroenterol. Hepatol. 2006, 4, 1403–1410.
- Alexander, J.; Kowdley, K.V. HFE-associated hereditary hemochromatosis. Genet. Med. 2009, 11, 307–313.
- Beutler, E. The significance of the 187G (H63D) mutation in hemochromatosis. Am. J. Hum. Genet. 1997, 61, 762–764.
- Fitzsimons, E.J.; Cullis, J.O.; Thomas, D.W.; Tsochatzis, E.; Griffiths, W.J.H.; on behalf of the British Society for Haematology. Diagnosis and therapy of genetic haemochromatosis (review and 2017 update). Br. J. Haematol. 2018, 181, 293–303.
- Hamdi-Roze, H.; Beaumont-Epinette, M.P.; Ben Ali, Z.; Le Lan, C.; Loustaud-Ratti, V.; Causse, X.; Loreal, O.; Deugnier, Y.; Brissot, P.; Jouanolle, A.M.; et al. Rare HFE variants are the most frequent cause of hemochromatosis in non-c282y homozygous patients with hemochromatosis. Am. J. Hematol. 2016, 91, 1202–1205.
- Merryweather-Clarke, A.T.; Cadet, E.; Bomford, A.; Capron, D.; Viprakasit, V.; Miller, A.; McHugh, P.J.; Chapman, R.W.; Pointon, J.J.; Wimhurst, V.L.; et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet. 2003, 12, 2241–2247.
- Wallace, D.F.; Subramaniam, V.N. Non-HFE haemochromatosis. World J. Gastroenterol. 2007, 13, 4690–4698.
- Bardou-Jacquet, E.; Ben Ali, Z.; Beaumont-Epinette, M.P.; Loreal, O.; Jouanolle, A.M.; Brissot, P. Non-HFE hemochromatosis: Pathophysiological and diagnostic aspects. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 143–154.
- Pietrangelo, A. Ferroportin disease: Pathogenesis, diagnosis and treatment. Haematologica 2017, 102, 1972–1984.
- Donker, A.E.; Raymakers, R.A.P.; Vlasveld, L.T.; Van Barneveld, T.; Terink, R.; Dors, N.; Brons, P.P.T.; Knoers, N.V.A.M.; Swinkels, D.W. Practice guidelines for the diagnosis and management of microcytic anemias due to genetic disorders of iron metabolism or heme synthesis. Blood 2014, 123, 3873–3886.
- Marchi, G.; Busti, F.; Lira Zidanes, A.; Castagna, A.; Girelli, D. Aceruloplasminemia: A Severe Neurodegenerative Disorder Deserving an Early Diagnosis. Front. Neurosci. 2019, 13, 325.
- Piperno, A.; Pelucchi, S.; Mariani, R. Inherited iron overload disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25.
- Gangaidzo, I.T.; Moyo, V.M.; Saungweme, T.; Khumalo, H.; Charakupa, R.M.; Gomo, Z.A.R.; Loyevsky, M.; Stearman, R.; La Vaute, T.; Enquist, E.G.; et al. Iron overload in urban Africans in the 1990s. Gut 1999, 45, 278–283.
- Gordeuk, V.R. African iron overload. Semin. Hematol. 2002, 39, 263–269.
- Siah, C.W.; Ombiga, J.; Adams, L.A.; Trinder, D.; Olynyk, J.K. Normal iron metabolism and the pathophysiology of iron overload disorders. Clin. Biochem. Rev. 2006, 27, 5–16.
- Bonkovsky, H.L.; Lambrecht, R.W.; Shan, Y. Iron as a co-morbid factor in nonhemochromatotic liver disease. Alcohol 2003, 30, 137–144.
- Sebastiani, G.; Walker, A.P. HFE gene in primary and secondary hepatic iron overload. World J. Gastroenterol. 2007, 13, 4673–4689.
- Tung, B.Y.; Emond, M.J.; Bronner, M.P.; Raaka, S.D.; Cotler, S.J.; Kowdley, K.V. Hepatitis C, iron status, and disease severity: Relationship with HFE mutations. Gastroenterology 2003, 124, 318–326.
- Ryan Caballes, F.; Sendi, H.; Bonkovsky, H.L. Hepatitis C, porphyria cutanea tarda and liver iron: An update. Liver Int. 2012, 32, 880–893.
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in fatty liver and in the metabolic syndrome: A promising therapeutic target. J. Hepatol. 2011, 55, 920–932.
- Rametta, R.; Dongiovanni, P.; Pelusi, S.; Francione, P.; Iuculano, F.; Borroni, V.; Fatta, E.; Castagna, A.; Girelli, D.; Fargion, S.; et al. Hepcidin resistance in dysmetabolic iron overload. Liver Int. 2016, 36, 1540–1548.
- Barisani, D.; Pelucchi, S.; Mariani, R.; Galimberti, S.; Trombini, P.; Fumagalli, D.; Meneveri, R.; Nemeth, E.; Ganz, T.; Piperno, A. Hepcidin and iron-related gene expression in subjects with Dysmetabolic Hepatic Iron Overload. J. Hepatol. 2008, 49, 123–133.
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634.
- Rostoker, G.; Griuncelli, M.; Loridon, C.; Couprie, R.; Benmaadi, A.; Bounhiol, C.; Roy, M.; Machado, G.; Janklewicz, P.; Drahi, G.; et al. Hemodialysis-associated hemosiderosis in the era of erythropoiesis-stimulating agents: A MRI study. Am. J. Med. 2012, 125, 991–999.e1.
- Kalantar-Zadeh, K.; Rodriguez, R.A.; Humphreys, M.H. Association between serum ferritin and measures of inflammation, nutrition and iron in haemodialysis patients. Nephrol. Dial. Transpl. 2004, 19, 141–149.
- Shenoy, N.; Vallumsetla, N.; Rachmilewitz, E.; Verma, A.; Ginzburg, Y. Impact of iron overload and potential benefit from iron chelation in low-risk myelodysplastic syndrome. Blood 2014, 124, 873–881.
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015, 126, 2031–2037.