Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Atorvastatin (ATS) is the gold-standard treatment worldwide for the management of hypercholesterolemia and prevention of cardiovascular diseases associated with dyslipidemia.
- atorvastatin
- lactonization
- active metabolites
- open acid form
1. Introduction
Statins are the first-line treatment of choice/gold-standard in the pharmacological management of hypercholesterolemia [1], and they have been positioned as the most effective oral drugs for the treatment and prevention of cardiovascular diseases associated with dyslipidemia [2,3]. Statins are reversible inhibitors of 3-hydroxy-3-methyl-glutharyl-coenzyme A (HMG-CoA) reductase, the enzyme responsible for de novo cholesterol biosynthesis. Statins can be administered in the active form (atorvastatin, fluvastatin, pitavastatin, pravastatin, and rosuvastatin) or as inactive drugs (simvastatin and lovastatin), which require activation within the organism. Statins are, in general, safe and well tolerated [4,5]. In terms of safety concerns, the most frequent adverse events are myopathies (5–10%) [6], ranging from muscle pain to very rare cases of fatal rhabdomyolysis [7], and hepatotoxicity, which is present in 1 % of treated patients and resolves spontaneously after withdrawal of the drug [5].
Among the statins, atorvastatin (ATS) is one of the most prescribed [8] statin worldwide for the treatment of hypercholesterolemia in order to diminish the cardiovascular risk [9]. ATS is a second-generation synthetic statin that is administered as the calcium salt of the active hydroxy-acid form, although some generics have been developed with the magnesium salt to avoid the patent protection of the calcium salt. According to the desired reduction in low-density lipoprotein cholesterol (LDLc) levels, the clinical posology involves the use of 10–80 mg once daily at any time of the day. Despite its wide, cost-effective use and pharmacological response, several factors undermine the clinical response of statins in the treatment of hypercholesterolemia, involving low adherence of patients, inadequate selection of the active ingredient, polymorphisms, adverse events (myopathies), drug–drug interactions (DDIs), etc. The use of model-based strategies able to properly characterize the time-course of the active entities is encouraged in order to optimize the dosing strategy in patients. Physiologically based pharmacokinetic (PBPK) modelling has emerged as a solid tool in the decision-making process during drug development, which has gained regulatory recognition in the last years [10,11]. The main applications of PBPK models range from drug–drug interactions (DDI), transporter evaluation, food–drug interactions, intrinsic factors evaluation, and extrapolation of drug exposure in special subgroups of patients.
2. Physicochemical Properties
2.1. Solubility
ATS (546 g/mol, pKa 4.46) belongs to class II of the Biopharmaceutical Classification System (BCS) due to its low solubility in gastrointestinal fluid, which contributes to its low bioavailability (12%) [12,13,14]. ATS solubility in deionized water is reported to be 0.0206 mg/mL at 37 °C [14]. A tri-hydrated calcium salt form of ATS (ATS-Ca) is included in the commercially available tablets of ATS. ATS-Ca has been isolated in amorphous and crystalline forms, but it is commercialized in its crystalline form due to the higher stability. ATS-Ca solubility increases with pH, being insoluble in acidic aqueous solutions of pH < 4 [15]. Solubility values in aqueous media for the amorphous and crystalline forms at 37 °C are 0.11–0.12 mg/mL in water, 0.01 mg/mL in HCl 0.1 N, and 0.72 and 0.70 mg/mL in sodium phosphate 0.05 M pH 7.4, respectively [16]. Great efforts have been performed to improve ATS oral bioavailability through formulation strategies to increase the solubility and/or dissolution rate of ATS-Ca such as micronization by antisolvent precipitation [15], microcapsulation [17], co-grinding techniques [18], co-amorphous formulations with nicotinamide [19], dry emulsions [13], inclusion complexes [20], and use of drug resinates [21]. Since ATS is administered mainly as the calcium salt, low solubility in the gastrointestinal (GI) tract should be considered in order to assess its PK characteristics.
2.2. Lipophilicity
The chemical structure of ATS (and of statins in general) can be divided into three parts: (1) the analogue of the target enzyme substrate (3-hydroxy-3-methyl-glutaril coenzyme A or HMG-CoA), (2) a complex hydrophobic ring structure covalently linked to the substrate analogue, and (3) side groups on the rings that define the solubility and PK properties [1]. While the analogue of the HMG-CoA (the mevalonate-like pharmacophore) is responsible for the reversible inhibition of the HMG-CoA reductase, the ring structure and its substituents lead to differences in lipophilicity, absorption properties, plasma protein binding, and elimination [22]. Lipophilicity of ATS is determined by its logP of 4.1 [22] and its logD at pH 7.4 (1.52) [3].
3. Absorption
A rapid oral absorption is expected after ATS administration, since the median Tmax is reported to be 1 h, with a range of 0.5–3 h [3]. The oral fraction absorbed of ATS is 30% between 10 and 80 mg, and its oral bioavailability is known to be low (12%) [22,23,24,25] and dose-independent. Therefore, dissolution and pre-systemic metabolism (gut wall and liver first-pass effect) are the key relevant processes affecting ATS bioavailability. The rate and extent of ATS absorption are influenced by the time of administration [26] and the presence of food [23]. A study with 16 healthy volunteers revealed that ATS maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) diminished by 47.9% and 12.7%, respectively, when an 80 mg dose was administered with food [27]. This reduction in ATS exposure has also been reported at the lowest dose level (10 mg) [28]. In this sense, the administration of ATS with food decreases its bioavailability by 13% [22,23]. Despite the administration of ATS in the evening is associated with a lower exposure compared to when it is dosed in the morning (with mean Cmax and AUC values 31% and 57% lower, respectively), and a food effect has been determined, no difference in the clinical response is observed [26,28]. For this reason, ATS can be administered at any time of the day and without regard to food. Gender is another covariate influencing ATS exposure, but it lacks any clinical relevance, despite the 10% lower AUC and 20% higher Cmax in females compared to males [12].
The intestinal absorption of ATS is a complex process, as the net transport of this drug through the gut wall involves multiple mechanisms, being not only restricted to passive diffusion. In vitro experiments in Caco-2 cell monolayers revealed an apparent permeability (Papp) in the basolateral-to-apical direction 7-fold higher than in the apical-to-basolateral direction, showing the role of P-glycoprotein (P-gp) efflux (Km and Jmax values of 115 ± 19 µM and 141 ± 11 pmol/cm2/min, respectively [29]). The interaction of ATS and P-gp has also been demonstrated in Madin–Darby canine kidney cells (MDCK) expressing human P-gp [30]. In this cell line, the efflux ratio after correcting with parental MDCKII cells resulted in 4.46 for ATS acid, suggesting ATS acid as a moderate substrate of P-gp. Moreover, monocarboxylic acid co-transporter (MCT) has been identified as a relevant transporter in the ATS absorption from the gut lumen with a Km value in the mM range. As clinically relevant maximal concentrations in the intestinal lumen are estimated to be within the 70–550 µM range after doses of 10 to 80 mg [3], ATS MCT-mediated absorption may be a linear process at this concentration, which is consistent with the proportional increase in the extent (AUC) of ATS absorption in the 10 to 80 mg dose range.
4. Distribution
The passive membrane permeability of statins increases along with their lipophilicity, and, consequently, lipophilic statins are distributed into peripheral tissues [31]. ATS has a volume of distribution of 5.4 L/kg [24] and exhibits a high degree of plasma protein binding (>98%) [32]. Statin accumulation in the liver is mediated by hepatic uptake through the organic anion transporting polypeptide (OATP) family, sodium-dependent taurocholate co-transporting polypeptide (NTCP), and by efflux transporters of the ATP-binding cassette (ABC) family, located on the basolateral and canalicular membranes of the liver, respectively [33]. In vitro kinetic studies on ATS hepatic uptake revealed that OATP1B1 and OATP1B3 were the major ATS uptake transporters, while NTCP was found to be of minor importance in ATS disposition. The average contribution to ATS uptake resulted as OATP1B1 > OATP1B3 >> OATP2B1 > NTCP; their respective Km (µM) values are 0.77, 0.73, 2.84, and 185 and Vmax (pmol/min/mg protein) values are 6.61, 2.29, 24.27, and 2260, respectively [34]. An ATS intrinsic uptake clearance of 2030 mL/min (95% CI: 1140–2620 mL/min) was predicted and, assuming the same passive diffusion across the cell membrane of hepatocytes and HEK293 cells (120 µL/min/g of liver), transporter-mediated active uptake of ATS dominates overall ATS hepatic uptake [34]. Moreover, polymorphisms in transporter genes have been reported to affect the PK of statins and their therapeutic effects [35,36]. It has been demonstrated that the liver-to-plasma concentration ratio of ATS is 2.7-fold higher (p = 0.002) in wild-type when compared to Slco1b2−/− mice after 1 mg/kg ATS tail vein injection [33]. In humans, it has been observed that ATS and its metabolites are sensitive to polymorphisms in SLCO1B1, as plasma concentrations were higher in subjects carrying the reduced function SLCO1B1 521C allele (T/C genotype) compared with the wild-type subjects (521 T/T) [37]. Another example comes from a fixed-order crossover study in 660 Finnish healthy volunteers [35], which concluded that individuals carrying the ABCG2 c.421C > A single-nucleotide polymorphism (SNP) had a 72% higher ATS AUC0-inf than individuals with the c.421CC genotype (p = 0.049), suggesting that the ABCG2 polymorphisms affect the PK of ATS. As the elimination half-life was not influenced by ABCG2 polymorphism, it allowed the authors to conclude that ABCG2 influences mostly during the absorption phase, enhancing ATS absorption and bioavailability [35].
5. Metabolism
Metabolism of ATS is an intricate pathway of different reactions that include glucuronidation [7,8,38], lactonization [39], and cytochrome P450-mediated oxidation [40,41]. A simplified scheme with the different metabolic pathways of ATS is depicted in Figure 1. ATS is administered as the hydroxy acid form (calcium salt), and its active metabolites (ortho-hydroxy atorvastatin (2OH-ATS) and para-hydroxy atorvastatin (4OH-ATS)) are equipotent to the parent compound, being responsible for 70% of the HMG-CoA reductase inhibitory activity of ATS [42]. The in vitro HMG-CoA reductase inhibitory activity (IC50) values for ATS, 2OH-ATS, and 4OH-ATS are 3.71, 5.54, and 3.29 nM, respectively [43]. Both metabolites, as the parent compound, are equilibrated with the corresponding lactone forms (ATS-L, 2OH-ATS-L, and 4OH-ATS-L) [38,39,41]. It has been demonstrated that lactonization might occur non-enzymatically at pH < 6 [44] or enzymatically, being the former pathway negligible at pH > 6. The formation of an acyl-glucuronide prior to lactonization is expected to be the major pathway for the enzymatic lactonization of ATS in humans, which is catalyzed by UDP-glucuronosyltransferases (UGTs) UGT1A1, UGT1A3, and UGT2B7. The isoenzyme UGT1A3 is the major contributor to this process with 200 times more activity than UGT2B7 [7]. The mechanism proposed for the lactonization is the formation of an acyl-β-D-glucuronide conjugate of the ATS acid (parent), elimination of the glucuronic moiety, and final spontaneous cyclization to the corresponding lactone [38]. ATS glucuronidation, and thus lactonization, follows non-linear kinetics with Km values of 4 and 20 µM and Vmax values of 2280 and 120 pmol/min/mg for UGT1A3 and UGT2B7, respectively [7]. ATS lactonization is affected by polymorphisms in the UGT1A locus and has been demonstrated both in vitro and in vivo in healthy volunteers [8]. On the other hand, the hydrolysis of the lactone forms of ATS and its metabolites to the corresponding carboxylates takes place non-enzymatically at pH > 6 [44] or can be catalyzed by plasmatic esterases or paraoxonases (PONs) [38]. PONs are a family of esterase/lactonase enzymes whose encoding genes are located in tandem in the long arm of human chromosome 7 (7q 21–22) [45], and PON1, PON2, and PON3 are highly involved in ATS metabolism. In addition, ATS increases the expression of PON2 [46]. A 3.8-fold higher ATS-L hydrolysis rate through PON1 and PON3 has been demonstrated in vitro when compared to spontaneous hydrolysis in a pooled microsomal fraction [47]. Additionally, results from incubation experiments in human liver microsomes (HLMs) show a median ATS formation rate through hydrolysis of the corresponding lactone of 309.70 pmol/min/mg protein [47]. Hydrolysis of lactone forms has been demonstrated to occur in plasma [48]. Therefore, this process must be considered when modelling ATS and its metabolites to better assess its pharmacokinetics.
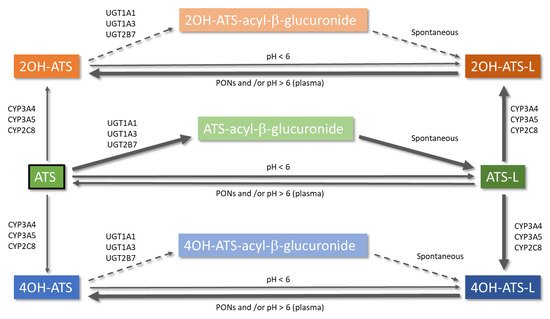
Figure 1. Metabolic pathways of atorvastatin. Arrow thickness informs directly about the relevance of the reaction and the sense of the equilibrium. Dashed arrows represent a theoretically possible lactonization of 2OH-ATS and 4OH-ATS via an acyl-β-glucuronide. ATS: atorvastatin open acid form; ATS-L: atorvastatin lactone form.
Cytochrome P450-mediated oxidative metabolism has been described as a major pathway of biotransformation for statins in humans [38], where CYP3A4 is the major enzyme involved in the formation of the two hydroxy-metabolites of ATS [39,41]. The CYP3A4-mediated oxidation is clearly polarized to the lactone forms, with Km values of 3.9 and 1.4 µM and Vmax values of 4235 and 14312 pmol/min/mg for the ortho- and para-hydroxylated metabolites, respectively [39]. Differences in Km and Vmax values between the acid and lactone form of ATS result in an intrinsic clearance ratio lactone/acid equal to 73 [40] and in specific metabolite clearance ratios for ortho-hydroxylation and para-hydroxylation of 20.2 and 83.1, respectively [39]. Quantum mechanics/molecular mechanics (QM/MM) have revealed that the acid form of ATS must pay a desolvation penalty of 5 Kcal/mol to enter in the more hydrophobic active site of the enzyme [39]. Moreover, the higher Vmax value for the para-hydroxylation of ATS-L has been attributed to a shorter distance to the heme oxygen atom of CYP3A4 [39]. Inhibition studies have demonstrated that ATS-L could be an inhibitor of the metabolism of the acid form [39]. It could be concluded that ATS lactonization changes its affinity to CYP3A, affecting the preferred hydroxylation positions, and may be responsible for DDIs at this level.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics13050709
This entry is offline, you can click here to edit this entry!