Genome editing in bacteria encompasses a wide array of laborious and multi-step methods such as suicide plasmids. The discovery and applications of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas based technologies have revolutionized genome editing in eukaryotic organisms due to its simplicity and programmability.
- CRISPR-Cas
- prokaryotes
- genome editing
- ribonucleoprotein
- suicide plasmids
1. Introduction
CRISPR (Clustered Regularly Inters ced Short Palindromic Repeats) is the only known adaptive and hereditary immune response in prokaryotes. It is present in about 50% of bacteria and 90% of archaea [1]. CRISPR-Cas acts by recompiling and storing genetic sequences from invader bacteriophages and noxious plasmids as spacers. These spacers are transcribed into crRNAs that bind to effector CRISPR nucleases (Cas proteins), which target specific complementary sequences, given they fulfill a specific PAM sequence requirement [2][3]. It was previously discovered that crRNAs need to couple to a RNAse III-edited tracRNA before binding to the Cas nuclease [4] (Figure 1). Depending on the number of effector proteins there are several types (I to VI) of CRISPR systems. Type II systems (e.g., Cas9) only depend on one effector nuclease which facilitates its heterologous expression, and are therefore the most popular tools for genome editing.
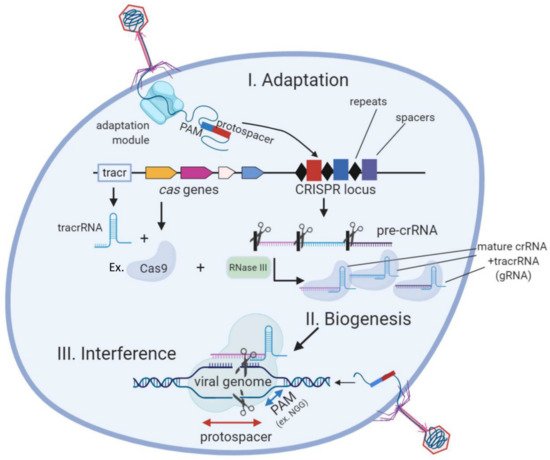
Figure 1. The three stages of the CRISPR-Cas (type II) bacterial adaptive immune system. During CRISPR adaptation, the injection of phage DNA into bacterial cell activates the adaptation module proteins which excise spacer-sized fragments of phage DNA for incorporation into CRISPR loci. During CRISPR RNA biogenesis, CRISPR loci are transcribed and resulting pre-crRNA is processed by a Cas9/RNaseIII complex at repeat sequences to generate mature crRNAs that couple to tracrRNA (gRNA). Individual gRNAs are bound by Cas protein effectors (e.g., Cas9). After a new phage infection with sequences matching a CRISPR spacer appears in the cell (lower right), specific Cas/gRNA complexes bind to viral DNA and cleave it.
2. Methods for Genome Editing in Prokaryotes
There are several methods that have been developed for genome editing in bacteria and are still widely used besides CRISPR-based tools. These methods, however, are highly laborious, often show inconsistent efficiencies, and require extensive tailoring for programming compared to simple gRNA design for CRISPR. Some of the most representatives are:
2.1. Suicide Plasmids
Suicide plasmids are those that can replicate in one organism, but not in another called recipient. These plasmids contain a homologous sequence (with the desired insertion, deletion or site-directed mutation) coupled to a marker, usually an antibiotic resistance cassette, and may harbor a transposon sequence that facilitates their insertion into the genome of the recipient strain after conjugation with the donor. As plasmid replication is not possible in the recipient strain or species, antibiotic treatment will select only those colonies that undergo genome integration (“Classical” method, Figure 2A). Suicide plasmids can incorporate an I-SceI site between the mutant allele and the antibiotic resistance marker. The suicide plasmid is transformed into an E. coli strain already harboring an inducible plasmid for I-SceI expression. The suicide plasmid is then integrated into the genome and colonies are selected by their antibiotic resistance at the non-permissive temperature for plasmid replication. Induction of I-SceI cleaves the target gene locus, which is then repaired via native RecA-mediated homologous recombination, providing large enough homology arms (>500 bp). This can result either in a reversion to the wild-type chromosome or in a markerless allele replacement [5] (“Scarless” method, Figure 2B).
2.2. Lambda Red System
Lambda Red consists primarily of three proteins: α, β, and γ. α is an exonuclease (exo), which processively digests the 5’-ended strand of a dsDNA end. β (bet) binds to ssDNA and promotes strand annealing. Finally, γ (gam) binds to the bacterial RecBCD enzyme (which degrades any linear DNA used as a template) and inhibits its activities. These proteins induce a “hyper-recombination” state in E. coli and other bacteria, in which recombination events between DNA species with as little as 35–50 bp of shared sequence occur at high frequency [6][7][8]. The system itself is however selection-free and therefore is usually combined with the insertion of large antibiotic-resistance cassettes to improve the recovery of edited colonies [6] (Figure 2C). In some cases, I-SceI sites are also included in the targeting construct to proceed with a counter-selection step to eliminate the resistance marker by homologous recombination [9].
2.3. ClosTron Method
The ClosTron system uses an element derived from the broad host range Ll.LtrB intron of Lactococcus lactis. Intron target specificity is determined by a small region, so it is cost-effective to re-target an intron by sub-cloning of a small DNA fragment (Figure 2D).
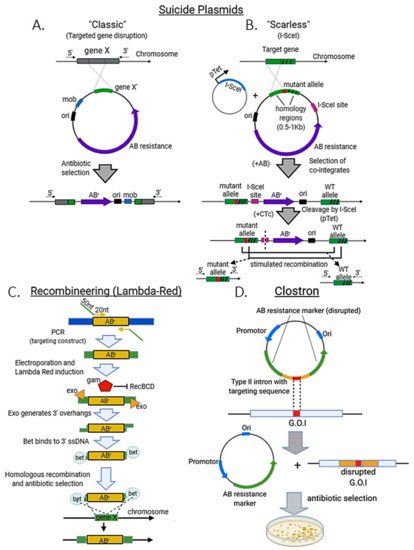
Figure 2. Standard methods for genome editing in bacteria. Suicide plasmids. (A) The classic approach consists in transforming with a non-replicating plasmid (usually with a transposon element, e.g., mob), which harbors a mutated recombination template and an antibiotic resistance marker (ABr). Antibiotic treatment will select only colonies that undergo homologous recombination to incorporate the plasmid sequence (including the mutant gene) at the target locus (disrupting gene X). (B) In the “scarless” variant a SceI site is incorporated in the plasmid to be transformed in a I-SceI expressing strain under an inducible promoter (pTet). After a first round of antibiotic treatment, cointegrating colonies harboring the plasmid sequence and the wild-type allele at the target gene locus are selected. Addition of chlortetracycline (CTc) induces I-SceI expression to cleave the target locus, which enhances homologous recombination to eliminate plasmid sequence resulting in either, reversion to wild-type or fixation of the mutant allele. (C) Recombineering (lambda red system) for targeted gene disruption. A targeting construct with 50 nt of homologous sequence at the 5′ and 3′ ends and antibiotic resistance marker is made by PCR. PCR template is electroporated and expression of the lambda Red proteins is induced (Ex. Heat shock at 42 °C). Gam inhibits RecBCD nuclease activity upon linear DNA (protecting the targeting construct). Exo generates 3′ overhangs in the DNA linear template, which are accessed by bet protein to facilitate homologous recombination and integration and disruption of the target gene (gene x). Edited colonies are then selected by antibiotic treatment. (D) ClosTron method. A type II intron with transposon activity is cloned within a disrupted antibiotic resistance cassette in a plasmid. After transformation, the intron, which has been modified with a specific, homologous sequence, targets the gene of interest (G.O.I) and disrupts it leaving behind a plasmid with a functional antibiotic resistance marker. Antibiotic selection then enhances and simplifies the obtention of mutant colonies.
3. CRISPR-Cas9 as a Genome-Editing Tool
Among the different Cas systems, the Cas9 protein from Streptococcus pyogenes (SpCas9) is currently the most widely used as a gene-editing tool. This is mostly due to its relatively common PAM sequence requirement: NGG (where N can be any nucleotide), with a theoretical frequency of once in every 8 bp in a random double-strand DNA sequence. The actual frequency of the PAM motif will vary across genomes and is expected to be much rarer in AT rich genomes. The other key element of the CRISPR-Cas system is the recombination template that contains flanking homology arms, the desired edit (insertion, deletion or specific mutation), and an internal sequence that disrupts the target site (e.g., mutations to the PAM), preventing targeting upon successful recombination.
4. CRISPR-Cas9-Based Methods for Genome Editing in Bacteria
Since its discovery, CRISPR Cas9 evolved as one of the main genome-editing tools in many organisms, including bacteria and a wide array of CRISPR-Cas9-based methods have been developed. These methods can vary on the number of plasmids used, the use of heterologous recombinase (e.g., lambda Red), and the DNA repair mechanism induced (e.g., HDR) (Figure 3).
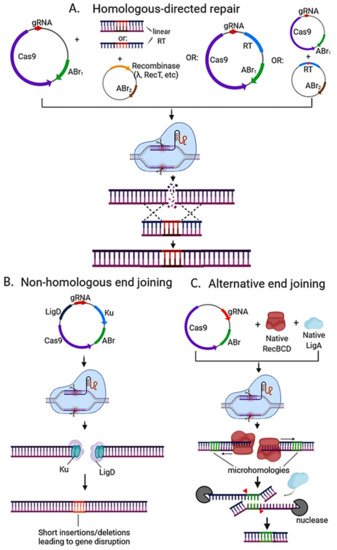
Figure 3. Strategies used for CRISPR-Cas based genome editing in bacteria. (A) Editing via homologous recombination: Recombineering with a linear DNA template is followed by counterselection with CRISPR nucleases. A heterologous recombinase (e.g., λ red, RecT) is introduced via a plasmid (or phage) into the cell and co-transformed with the linear DNA template and CRISPR-nuclease plasmid with respective antibiotic-resistance marker (ABr). Genome editing may also be directed with a plasmid-encoded recombination template (RT) and endogenous or heterologous recombinase. The recombination template can be placed on the same plasmid encoding the CRISPR machinery for an all-in-one plasmid system, or it can be placed on a separate plasmid before transforming the CRISPR nuclease/gRNA plasmid. One-plasmid system is more streamlined, but due to its larger size it can be hard to transform, and cloning may not be possible if the gRNA can target the genome of the cloning strain. (B) Editing via the non-homologous end-joining (NHEJ) pathway. Depending on the strain, ku and/or ligD can be encoded on the CRISPR nuclease/gRNA plasmid and transformed into the strain. (C) Alternative end joining (A-EJ) pathway can be found natively in many bacterial species with incomplete NHEJ. It does not require the introduction of foreign Ku or LigD, and instead relies in microhomology-directed repair via RecBCD, nucleases, and LigA, leading to deletions of variable sizes (depending on the location of microhomologies) at the Cas9 cut site. For a more detailed insight on NHEJ and A-EJ mechanisms, the reader is advised to read [10]. All strategies require plasmid curing after nuclease targeting to isolate the mutant strain in order to avoid interference in pursuing downstream applications.
5. Alternatives to SpCas9-Associated Cytotoxicity and Lack of Colonies: Expanding the Toolbox
SpCas9 has been used almost exclusively to perform genome editing in bacteria since its original application in E. coli [11]. This is mostly due to its relatively simple PAM sequence requirement, but also to its well-characterized crystal structure and molecular mechanism of action (Figure 4). In bacteria where there is also a poor transformation efficiency and/or weak DNA repair mechanisms, these effects sum up and can turn into no colonies when the system is used for genome editing. To ameliorate these issues, inducible promoters have been used to drive SpCas9 expression (Figure 5A). SpCas9-RuvC domain has been mutagenized (D10A) to function as a DNA nickase to produce single-strand breaks instead of the more lethal DSB (Figure 5B).
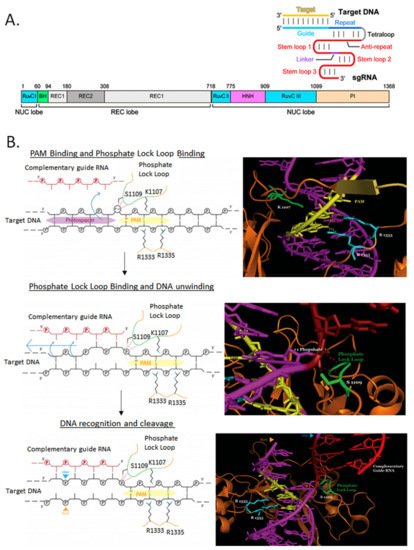
Figure 4. Molecular mechanism of SpCas9/gRNA cleavage of target DNA. (A) The main domains of Cas9 are illustrated next to a gRNA/target DNA secondary structure scheme. Adapted from [12]. (B) The first step is the PAM binding and phosphate lock loop binding, followed by DNA unwinding and finally the DNA recognition by gRNA and the target DNA cleavage by the RuvC and HNH nuclease domains at both strands. Critical Cas9 residues for each step are illustrated. Adapted from [13].
One of the most recent advances in genome editing are the base editors, which specifically perform single-nucleotide edits without a double strand break or recombination template. The most extended base editor, BE3, is composed of a chimera of nCas9 to provide strong, specific gRNA-programmable DNA binding, and cytidine-deaminases, e.g., APOBEC1, to conduct C to T editing in the target gene [14] (Figure 5C). Other variants of the system include an adenine-deaminase (A to G conversion) instead of a cytidine-deaminase [15]. This system was initially developed in eukaryotes but is becoming more common in some bacteria like E. coli [16], Klebsiella pneumonia [17], Pseudomonas aeruginosa [18], Rhodobacter sphaeroides [19], and Staphylococcus aureus [20]. The advantages of these systems include its relative innocuity compared to Cas9-induced DSBs, and its independence from recombination machinery to introduce specific single-nucleotide mutations in a target gene.
Although base-editors are optimized for single-base edits, replacing larger stretches of genomic DNA by inserting sequences such as an epitope tag or a deletion usually requires a foreign DNA donor to repair a Cas9-induced DSB. A type V-K CRISPR-associated transposase (ShCAST) system avoids these requirements (Figure 5D). This method is based on a naturally occurring Tn7-like transposon from Scytonema hoffmani which encodes besides its transposase genes, a nuclease-deficient Cas12k, tracRNA and 28–34 bp crRNAs [21]. ShCAST transposases, Cas12k and targeting sgRNAs are cloned into a helper plasmid, while cargo genes flanked by LE and RE elements to facilitate their insertion into a crRNA-targeted locus, are cloned into a donor plasmid. Integration is not “scarless” as it also includes the LE and RE elements and a 5-bp duplication at the insertion site. The ShCAST system has shown up to 80% genome editing efficiency in several E. coli target loci without positive selection [21], highlighting its potential for genome engineering in prokaryotes.
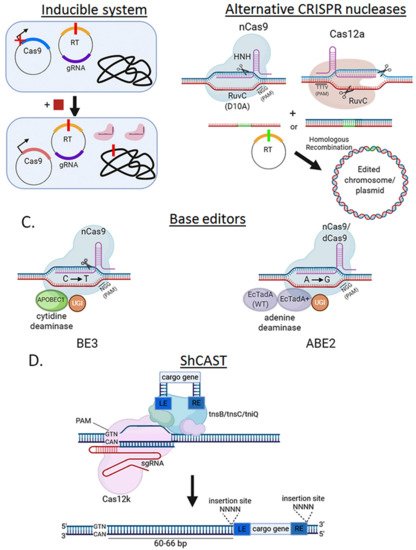
Figure 5. Alternative strategies to circumvent SpCas9 cytotoxicity. (A) Use of inducible systems to express SpCas9. Via an inducible promoter, SpCas9 expression is strongly repressed without inducer present (square) and only induced after exponential culture so that enough cells can survive and perform the genome edit. (B) Using less toxic nucleases to achieve editing. nCas9, which only cleaves one strand of DNA, and Cas12a (PAM: TTTV, where V is A or C or G) can be less toxic than SpCas9. (C) SpCas9-derived base editors eliminate the double-stranded break requirement for genome editing. A translational fusion of nCas9 (nickase) or dCas9 (“dead”), a cytidine (e.g., APOBEC1 in BE3) or adenosine (e.g., TadA-EcTadA+ in ABE2) deaminase domain, and an uracil DNA glycosylase inhibitor (UGI) is introduced on a plasmid into the cell. Upon nuclease binding and DNA strand unwinding, cytidines (or adenines) on the non-target strand within a defined window adjacent to the PAM are rapidly converted to uracil (or inosines), which is then processed as thymidine (or guanines) by DNA polymerase. (D) ShCAST insertion mechanism. A Tn7-like transposon from Scytonema hoffmani encodes transposases (tnsB, tnsC, tniQ), a nuclease deficient type V CRISPR protein (Cas12k) and guide RNA. This complex is combined with a cargo gene flanked by LE and RE elements. ShCAST is directed to the target locus and integrates the cargo gene 60–66 bp downstream of the PAM sequence, generating and insertion of the cargo gene flanked by the SE and RE elements, and a duplicated (4 bp) insertion site.
6. Summary
CRISPR-Cas technology has revolutionized the genome editing and has become the state-of the-art approach in eukaryotic organisms. Its application in prokaryotes has been slower but is quickly being adapted to several bacteria of industrial and biomedical importance. In particular, if it is to be superior to current methods based on suicide plasmids and recombineering that keep being adapted and improved in bacteria [22], efficiency needs to be adjusted regarding available transformation tools and genetic accessibility for each species. However, the continuous expression of any foreign protein with DNA-binding/editing activity seems to be particularly toxic for many bacteria. In eukaryotic organisms the ribonucleoprotein (RNP) format with foreign but well characterized Cas enzymes, such as SpCas9, has shown higher efficiency and much lower cytotoxic and off-target effects compared to the plasmids. Ultimately, genome editing would allow the creation of synthetic genomes combining a wide array of genes, metabolic pathways, and even full chromosomes [23] from different organisms to optimize the production of relevant metabolites, e.g., natural products [24].
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms9040844
References
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259.
- Giedrius, G.; Rodolphe, B.; Philippe, H.; Virginijus, S. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607.
- Fehér, T.; Karcagi, I.; Gyorfy, Z.; Umenhoffer, K.; Csörgo, B.; Pósfai, G. Scarless engineering of the Escherichia coli genome. Methods Mol. Biol. 2007, 416, 251–259.
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223.
- Yu, D.; Ellis, H.M.; Lee, E.-C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5978–5983.
- Poteete, A.R. What makes the bacteriophage λ Red system useful for genetic engineering: Molecular mechanism and biological function. FEMS Microbiol. Lett. 2001, 201, 9–14.
- Tas, H.; Nguyen, C.T.; Patel, R.; Kim, N.H.; Kuhlman, T.E. An integrated system for precise genome modification in Escherichia coli. PLoS ONE 2015, 10, e0136963.
- Chen, W.; Zhang, Y.; Yeo, W.-S.; Bae, T.; Ji, Q. Rapid and Efficient Genome Editing in Staphylococcus aureus by Using an Engineered CRISPR/Cas9 System. J. Am. Chem. Soc. 2017, 139, 3790–3795.
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239.
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014.
- Cavanagh, P.; Garrity, A. “CRISPR Mechanism”, DNA Binding and Cleavage. Available online: (accessed on 5 February 2021).
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424.
- Nicole M. Gaudelli; Alexis C. Komor; Holly A. Rees; Michael S. Packer; Ahmed H. Badran; David I. Bryson; David R. Liu; Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464-471, 10.1038/nature24644.
- Satomi Banno; Keiji Nishida; Takayuki Arazoe; Hitoshi Mitsunobu; Akihiko Kondo; Deaminase-mediated multiplex genome editing in Escherichia coli. Nature Microbiology 2018, 3, 423-429, 10.1038/s41564-017-0102-6.
- Yu Wang; Shanshan Wang; Weizhong Chen; Liqiang Song; Yifei Zhang; Zhen Shen; Fangyou Yu; Min Li; Quanjiang Ji; CRISPR-Cas9 and CRISPR-Assisted Cytidine Deaminase Enable Precise and Efficient Genome Editing inKlebsiella pneumoniae. Applied and Environmental Microbiology 2018, 84, e01834-18, 10.1128/aem.01834-18.
- Weizhong Chen; Ya Zhang; Yifei Zhang; Yishuang Pi; Tongnian Gu; Liqiang Song; Yu Wang; Quanjiang Ji; CRISPR/Cas9-based Genome Editing in Pseudomonas aeruginosa and Cytidine Deaminase-Mediated Base Editing in Pseudomonas Species. iScience 2018, 6, 222-231, 10.1016/j.isci.2018.07.024.
- Yufeng Luo; Mei Ge; Bolun Wang; Changhong Sun; Junyi Wang; Yuyang Dong; Jianzhong Jeff Xi; CRISPR/Cas9-deaminase enables robust base editing in Rhodobacter sphaeroides 2.4.1.. Microbial Cell Factories 2020, 19, 93-14, 10.1186/s12934-020-01345-w.
- Ya Zhang; Hongyuan Zhang; Zhipeng Wang; Zhaowei Wu; Yu Wang; Na Tang; Xuexia Xu; Suwen Zhao; Weizhong Chen; Quanjiang Ji; et al. Programmable adenine deamination in bacteria using a Cas9–adenine-deaminase fusion. Chemical Science 2020, 11, 1657-1664, 10.1039/c9sc03784e.
- Jonathan Strecker; Alim Ladha; Zachary Gardner; Jonathan L. Schmid-Burgk; Kira S. Makarova; Eugene V. Koonin; Feng Zhang; RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019, 365, 48-53, 10.1126/science.aax9181.
- Volke, D.C.; Friis, L.; Wirth, N.T.; Turlin, J.; Nikel, P.I. Synthetic control of plasmid replication enables target- and self-curing of vectors and expedites genome engineering of Pseudomonas putida. Metab. Eng. Commun. 2020, 10.
- Wang, K.; de la Torre, D.; Robertson, W.E.; Chin, J.W. Programmed chromosome fission and fusion enable precise large-scale genome rearrangement and assembly. Science 2019, 365, 922–926.
- Xu, X.; Liu, Y.; Du, G.; Ledesma-Amaro, R.; Liu, L. Microbial Chassis Development for Natural Product Biosynthesis. Trends Biotechnol. 2020, 38, 779–796.