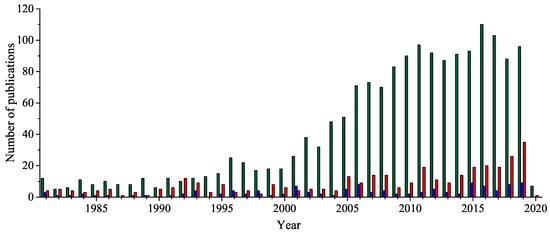
In the last 40 years, 1,2,4-oxadiazole heterocycle has been widely explored bringing a vast number of compounds exhibiting diverse biological activities such as anticancer, anti-inflammatory, anticonvulsant, antiviral, antibacterial, antifungal, antidepressant, antiangiogenic, analgesic, anti-insomnia, anti-oedema, antiparasitic, and anti-Alzheimer. It was proved that they also show inhibitory potency against Human Deacetylase Sirtuin 2 (HDSirt2), Carbonic Anhydrase (CA), Histone Deacetylase (HDAC), Rearranged during Transfection (RET) kinase, Penicillin-Binding Protein (PBP2a), efflux pump, cyclooxygenases (COX-1 and COX-2) and butyrylcholinesterase (BChE) as well as affinity to σ
1, σ
2, orexin, kappa opioid (KOR) and estradiol (ER) receptors (see sections below). Furthermore, 1,2,4-oxadiazole derivatives also found application as supramolecular liquid crystals and HEDMs [
7,
17,
18,
19]. Importantly, the heterocycle demonstrates bioisosteric equivalence with ester and amide moieties due to the possibility of creation specific interaction (e.g., hydrogen bonding). It is a particularly useful alternative when the instability of those groups is observed (e.g., when the hydrolysis may appear) [
20,
21]. Nowadays, there are a few commercially available drugs containing 1,2,4-oxadiazole nucleus such as
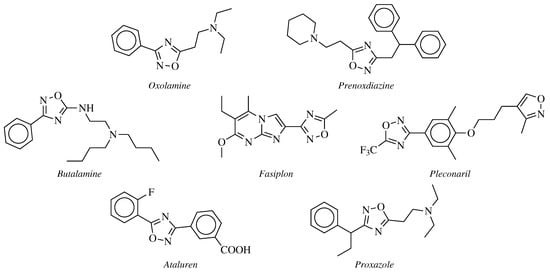
Oxolamine,
Prenoxdiazine (cough suppressant, )
Butalamine (vasodilator, ),
Fasiplon (nonbenzodiazepine anxiolytic drug, ),
Pleconaril (antiviral drug, ),
Ataluren (Duchenne muscular dystrophy treatment drug, ) and
Proxazole (a drug used for functional gastrointestinal disorders, ) [
22,
23,
24]. It is worth noting that 1,2,4-oxadiazole ring, as the only one of all oxadiazole isomers, occurs in the structures of natural products. For example, in 2011 Carbone M. et al. isolated two indole alkaloids
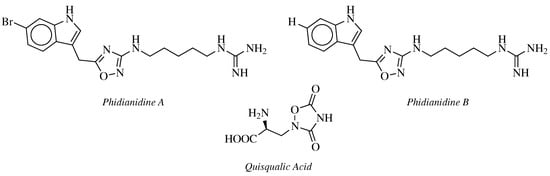
Phidianidine A and
Phidianidine B () from sea slug Opisthobranch
Phidiana militaris [
25].
3. Anticancer Agents
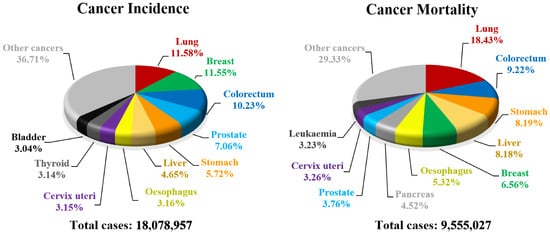
Every year cancer impacts about 20 million people all over the world resulting in deaths counting in millions (). Unfortunately, a number of new cancer cases is still rising and almost 30 million people will be diagnosed with carcinoma by 2040 in high-developed countries [
50]. For that reason, finding new cancer treatments or effective drugs is one of the greatest needs of the current community and a challenge for modern medicine. Biological evaluation of 1,2,4-oxadiazoles revealed that some of their derivatives are potent anticancer agents. The greatest breakthrough came with the discovery of 3,5-diarylsubstituted derivatives of 1,2,4-oxadiazole as a new series of apoptosis inducers [
51]. Since then, exploration of the anticancer activity of 1,2,4-oxadiazole derivatives has been started resulting in a creation of a wide library of compounds [
52,
53].
Figure 5. Estimated number of cancer incidences and cancer-related deaths in 2018.
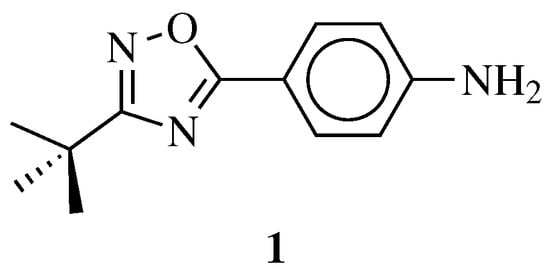
Recently, Maftei C. V. et al. reported the synthesis of 4-(3-(
tert-butyl)-1,2,4-oxadiazol-5-yl)aniline (
1, ), which exhibits moderate activity with a mean IC
50 value of approximately 92.4
μM against panel of 11 cancer cell lines (human colon adenocarcinoma—CXF HT-29, human gastric carcinoma—GXF 251, human lung adenocarcinoma—LXFA 629, human non-small cell lung carcinoma—LXFL 529, breast cancer-derived from athymic mice’ lung metastatic site—MAXF 401, human melanoma—MEXF 462, human ovarian adenocarcinoma—OVXF 899, human pancreatic cancer—PAXF 1657, human pleuramesothelioma cancer—PXF 1752, human renal cancer—RXF 486, human uterus carcinoma—UXF 1138). Importantly, compound
1 became a precursor for synthesis of novel compounds with greater antiproliferative activities [
54].
Figure 6. Chemical structure of 4-(3-(tert-butyl)-1,2,4-oxadiazol-5-yl)aniline 1.
Further modification of
1 led to the discovery of its derivative
2 () exhibiting significantly greater antitumor activity evaluated against a panel of 12 human tumor cell lines (CXF HT-29, GXF 251, LXFA 629, LXFL 529, MAXF 401, MEXF 462, OVXF 899, PAXF 1657, human prostate cancer—PRXF 22Rv1, PXF 1752, RXF 486 and UXF 1138), especially toward OVXF 899 and PXF 1752 cell lines with the IC
50 values of 2.76 and 9.27
μM, respectively. Moreover, compound
2 showed high selectivity against renal cancer cell line with the IC
50 = 1.143
μM [
55]. In addition, the same research team reported new gold(I) complexes with 1,2,4-oxadiazole-containing
N-heterocyclic carbene ligands. Obtained results clearly revealed impressive potency of imidazolium salts. The most active derivative
3 () showed extremely low IC
50 values from 0.003 to 0.595
μM against the same panel of 12 cancer cell lines with highest activity in an in vitro assays with LXFA 629 and MAXF 401 cells (IC
50 = 0.003
μM for both of them) [
56]. Thus,
3 seems to be an ideal candidate for further evaluation. More advanced in vivo studies may reveal some additional features, although no information has been published up to date.
Table 2. 1,2,4-oxadiazole derivatives with antitumor activity.
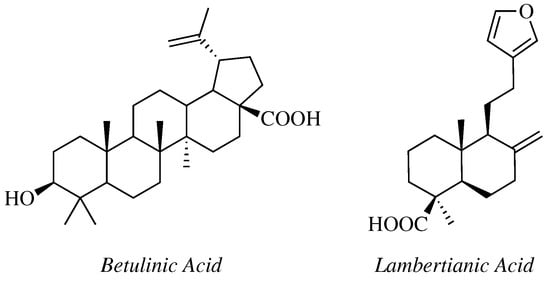
In a study reported by Challa K., Krishna C. and coworkers C28-modified
Betulinic Acid () bearing 1,2,4-oxadiazole ring connected via ester or amide linker have been synthesized and evaluated against human colon carcinoma (Colo 205), human liver cancer (Hep G2) and HeLa cell lines [
57,
58]. Performed screening revealed moderate potential of all obtained derivatives with the highest biological activities for analogs
4a–4d () (the IC
50 values in a range of 26.1–34.3
μM). However, the obtained compounds turned out to be still weaker than reference compound—etoposide (the IC
50 values of 0.42–22.5
μM), a topoisomerase II enzyme inhibitor, which is currently used as medication in the treatment of cancer diseases (e.g., lung, ovarian, testicular cancers, leukemia, neuroblastoma and lymphoma) [
59]. Interestingly, the impact on the biological activity of compounds by switching the ester moiety with amide was negligible.
Figure 7. Chemical structure of Betulinic Acid and Lambertianic Acid.
Mironov et al. carried out a synthesis of several derivatives of
Lambertianic acid () by the introduction of substituted-1,2,4-oxadiazole heterocycle at the C16 position [
60]. Obtained compounds were tested in comparison with doxorubicin—widely used anticancer agent in the treatment of breast, bladder carcinomas, lymphoma, and acute lymphocytic leukemia [
61,
62]. Obtained outcomes by Mironov et al. revealed that
5a–
b () exhibited more favorable biological activity than
Lambertianic Acid itself with the GI
50 values at sub-micromolar concentration against human childhood and adult T acute lymphoblastic leukemia (CEM-13), MT-4, and human adult acute monocytic leukemia (U-937) cancer cell lines. It is worth noting that
5a–
b demonstrated greater cytotoxic activity than doxorubicin. Additional biological studies indicated that activities of
5a–
b against human breast adenocarcinoma—MCF-7, MDA-MB-231 and human melanoma—MEL-8 cancer cell lines were slightly lower than the reference compound. Interestingly, flow cytometry assay revealed that the above-mentioned compounds are potent inducers of apoptosis in MCF-7, MDA-MB-231 and MEL-8 cell lines and are acting in a dose-dependent manner.
In the study of Kucukoglu K. et al. a series of Schiff bases fused with 1,2,4-oxadiazole heterocycle has been synthesized and evaluated in vitro against a panel of 8 cancer cell lines [
63]. Results revealed that
6a–
c () exhibited higher biological potency (CC
50 = 137.3, 79.0 and 140.3
μM, respectively) against Ca9-22 cell line than 5-fluorouracil (a multi-acting agent used in the treatment of colon, esophageal, stomach, breast and pancreatic cancers) applied as a reference (CC
50 = 214.3
μM). On the other hand, the cytotoxic potency of obtained compounds occurred to be far weaker than doxorubicin. For this reason, modifications of chemical structure including a different substitution of terminal aromatic rings or an introduction of additional pharmacophores are worth of consideration to improve biological activity.
Moniot S., Forgione M. et al. reported a study of about 40 novel substituted 3-aryl-5-alkyl-1,2,4-oxadiazole derivatives as selective inhibitors of HDSirt2—NAD
+ lysine deacetylase—an attractive target for treating neurodegenerative disorders, metabolic dysfunctions, age-related diseases and cancer [
64]. The biological activity of obtained derivatives was assessed in a continuous assay using an
α-tubulin-acetylLys40 peptide as a substrate. Based on the detailed structure-activity relationship (SAR) studies, compounds
7a and
7b () emerged as the most potent HDSirt2 inhibitors when tested against human leukemia cell lines (U-937, NB4, HL-60, and K562) and MDA-MB-231 cell line. Analog
7a was able to induce apoptotic death in over 80% of NB4, K562 and MDA-MB-231 cancer cell at the concentration of 25
μM. Moreover,
7b achieved the same effect at 10
μM. According to the western blot analyses, the involvement of HDSirt2 inhibition for apoptotic death induction has been confirmed. In addition, the crystal structure of 1,2,4-oxadiazole derivatives in complex with HDSirt2 revealed yet unexplored subcavity, which may be extremely useful for further inhibitors development [
64].
In 2017, Avanzo R. E. and coworkers synthesized 9 novel diheterocyclic-ribose fused derivatives containing 5-substituted-1,2,4-oxadiazole framework. Their previous study suggested that 5-deoxy-5-
S-(1,2,4-triazol-3-yl)-2,3-
O-cyclopentylidene-
β-D-ribofuranoside derivatives are moderate antitumor agents. It turned out that the introduction of 5-substituted-1,2,4-oxadiazole heterocycle into the ribose-derivative structure improved anticancer activity [
65,
81]. Obtained compounds were tested against human lung (A549), SW1573, HeLa, human breast (HBL-100), T-47D, and human colon (WiDr) cancer cell lines. Among them, compound
8 () showed the highest antiproliferative potency and selectivity against WiDr with the GI
50 value of 4.5
μM. It was noticed that the presence of electron withdrawing group (EWG) at the
para position of the aromatic ring occurred to be crucial to ensure high biological activity.
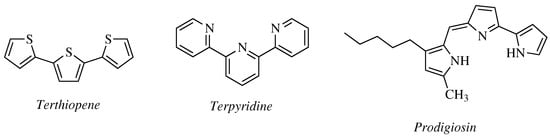
Recently, Abd el hameid M. K. reported 15 novel 1,2,4-oxadiazole derivatives as analogs of
Terthiopene,
Terpyridine, and
Prodigiosin ()—naturally occurring compounds with potent cytotoxic and pro-apoptotic properties against various types of carcinoma [
66]. Obtained compounds were preliminary evaluated against MCF-7 cancer cell line and the most potent were selected for further evaluation toward human colon cancer—HCT-116 cell line. Obtained results revealed that
9a–
c () exhibited the highest activity with the IC
50 values of 0.48, 0.78, 0.19
μM and 5.13, 1.54, 1.17
μM against MCF-7 and HCT-116, respectively. In addition, their biological activities were comparable or greater than reference
Prodigiosin (the IC
50 values of 1.93 and 2.84
μM against MCF-7 and HCT-116 cell line, respectively). Interestingly, flow cytometry analysis revealed that the above-mentioned compounds were able to arrest cell proliferation at G1 phase in MCF-7 cells and were triggering apoptosis via increasing of caspase3/7 activity, thus are suitable for further development as potent anticancer agents.
Figure 8. Chemical structure of Terthiopene, Terpyridine, and Prodigiosin.
4. Antimicrobial Agents
So far literature have listed over 1400 different species of microbials (including bacteria, viruses, protozoa, fungi and helminthes) able to elicit illnesses in human body which very often leads to death. Surprisingly, only 20 of them (mainly bacteria) are responsible for approximately two thirds of the fatal cases [
93]. Estimated deaths from infections is continuously falling, from 16 million in 1990 to approximately 15 million to forecasting 13 million in 2050 in high-developed countries. However, people are still suffering an enormous burden dint of pneumonia, HIV/AIDS, tuberculosis, malaria, diarrhea and many other diseases [
94,
95]. In light of the numerous pandemic threats in European countries and the world, including the recent infections with the SARS-CoV-2 virus causing COVID-19, discovering new, effective antibacterial/antiviral drugs and the development of modern therapies are two challenges of paramount importance.
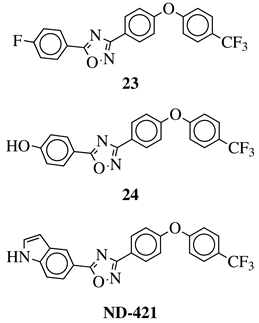
In 2014 O’Daniel P. I., Mobashery S., and Chang M. et al. from the University of Notre Dame in the United States put a great effort into the development of 1,2,4-oxadiazole as new antibiotics and discovered a new class of non-
β-lactam drugs that were able to inhibit PBP2a of Methicillin-Resistant
Staphylococcus aureus (MRSA) [
96]. Detailed computer screening allowed to select 29 compounds from 1.2 million compounds (ZINC database), which were tested for their antibacterial activity against ESKAPE pathogens and agent
23 () emerged as the most promising. Its further evaluation brought an enormous number of derivatives and led to the discovery of
24 (), which exhibited superior antibacterial activity against Vancomycin-Resistant
S.
aureus (VRSA), Vancomycin-Resistant
Enterococcus faecium (VRE) as well as MRSA with the MIC values ranging from 1 to 2
μg/mL. Moreover, rapid-time kill kinetics studies revealed that
24 was able to cause instant cell death of VRE and Daptomycin-non-Susceptible isolates at 4 mg/L in 1 h resulting in better outcomes than reference compound—daptomycin [
97]. Further modifications of
24 and very detailed SAR analysis allowed to obtain a wide library of its analogs (counting in hundreds of derivatives) and resulting in discovery of 5-(1
H-indol-5-yl)-3-(4-(4-(trifluoromethyl)phenoxy)phenyl)-1,2,4-oxadiazole (also called as
ND-421, ).
ND-421 exhibited longer half-time, a high volume of distribution, low clearance, excellent bioavailability, 3 times longer postantibiotic effect than linezolid without inoculum effect with unaltered biological activity [
98,
99,
100]. Additionally, in vitro studies against
S.
aureus, which exhibits two- and four-fold increased resistance, revealed first-time-reported, unique resistance mechanism to 1,2,4-oxadiazoles in MRSA. Moreover, those pathogen mutants did not show increased resistance to ampicillin, imipenem, linezolid, and vancomycin antibiotics (which are last drug-based defense against MRSA and VRSA) which made
ND-421 a perfect alternative drug for refractory microorganisms [
101]. It is also worth pointing out that
ND-421 showed high synergy with other
β-lactams (oxacillin, piperacillin, imipenem, meropenem and cefepime) unlike to non-
β-lactam antibiotics (vancomycin, linezolid, gentamicin, doxycycline and azithromycin). Recently, the same research team performed additional in vitro studies of
ND-421 against 210 different MRSA and VRE, which exhibited the MIC
50 values of 4
μg/mL in all examined strains. Moreover value of MIC
50 were consistently lowered when studied compound was used in combination with oxacillin [
102,
103]. In summary, 1,2,4-oxadiazoles
23,
24 and
ND-421 are extremely potent and very promising non-
β-lactam bactericidal antibiotics against Gram-positive multi-resistant bacteria suitable for further in vivo evaluation and clinical studies, although no information has been published up to date.
Table 3. 1,2,4-oxadiazole derivatives and their antimicrobial activity.
| General Structure |
Substituents |
The Most Active Derivatives |
Activity |
Ref. |
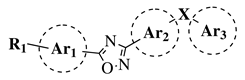 |
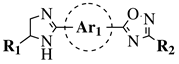
R1 = H, OH, OCH3, NH2, NHAc, NH3Cl, NHMs, NH-nBu, NH-tBu, NHCOPh, NH-iPr, PO3H2, PO(OEt)2, SO2NH2, CONH2, COOH, COOCH3 F, Cl, Br, I, NO2, ethynyl or CN;
Ar1 = phenyl, benzyl, 2-pyrole, 3-pyridyl, 4-pyridyl, 5-indole, 3-pyrrazole, 2-imidazole and many others (see Ref.);
Ar2, Ar3 = p-phenylene, 6-indole, 2-pyridyl, 6-chromene, carbazole, N-phenylpiperazine, N-phenylmorpholine and many others (see Ref.);
X = NH, CH2, O, CO, NBn, SO or SO2. |
 |
MIC50 values <4 μg/mL against over 210 diverse, MRSA and VRE strains. |
[96,98,99,103] |
 |
X = NH or none;
R1 = H, 3-chloro-4-fluorophenyl, 2-chlorophenyl, 2-ethyl, 4-ethyl, 5-bromo-2-fluorophenyl or 2-methylpyridin-5-yl. |
 |
Grown inhibition zone within 20–25 mm against S. aureus, B. subtilis, E. coli, P. vulgaris, P. aeruginosa, C. albicans. |
[104] |
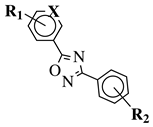 |
R1 = H, 2-chloro or 3-chloro;
X = CH or N;
R2 = H, 2-nitro, 2-chloro, 3-bromo, 2-chloro-5-nitro, 2-bromo, 3-nitro, 2-iodo, 3,5-dinitro, 4-nitro or 2-hydroxy. |
 |
MIC value of 60 μM against E. coli. |
[105] |
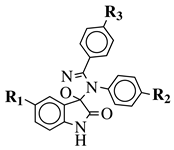 |
R1 = H, F, Cl, Br, I, methyl, ethyl, methoxy or iPr;
R2 = H, methyl, methoxy, iPr, F, Cl, Br or I;
R3 = H, F, Cl, Br, nitro, iPr, OBn, methoxy, ethoxy or CN. |
 |
MIC value of 64 μg/mL against S. epidermidis. |
[106] |
 |
R1 = H or methyl;
Ar1 = p-phenylene or m-phenylene;
R2 = methyl, cyclopropyl, 2-thienyl, 2-chlorophenyl, 3-chlorophenyl, 3,4-dichlorophenyl, 4-ethylphenyl, 4-t-butylphenyl, 4-methylphenyl, 3,4,-dimethylphenyl and many others (see Ref.). |
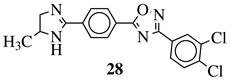 |
MIC values in a range 8–16 μg/mL toward S. aureus, B. subtilis, E. coli, P. fluorescent. |
[107] |
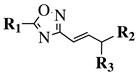 |
R1 = phenyl, 4-mehtoxyphenyl, 4-chlorophenyl, 3-methylthienyl or 2-pyridinyl;
R2, R3 = H, methyl, phenyl, 4-chlorophenyl, 4-methoxyphenyl, 3,4,-dimethoxyphenyl or 2,3-dimethoxyphenyl. |
 |
MIC value of 0.68 mM against S. aureus. |
[108] |
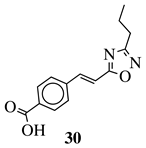
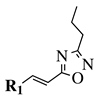 |
R1 = phenyl, 4-methylphenyl, 4-methoxyphenyl, 4-methylthiophenyl, 2-chlorophenyl, 4-chlorophenyl, 2-3-dichlorophenyl, 3,4-dichlorophenyl, 4-fluorophenyl, 4-bromophenyl, 4-hydroksyphenyl, 2-bromo-4-fluorophenyl, 4-cyanophenyl, 4-pyridinyl, 1-napthyl and others (see Ref.). |
 |
IC50 value of 0.045 μg/mL against M. tuberculosis (H37Ra). |
[109] |
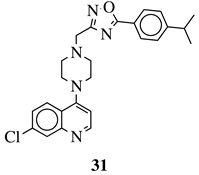
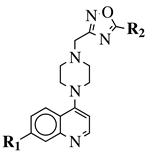 |
R1 = 4-pyridyl, 3-pyridinyl or 3,5-difluorophenyl;
R2 = 3,5-dimethoxyphenyl, 3,5-difluorophenyl, 3-cyanophenyl, 2,3-dimethylphenyl, cyclopentyl or 4-izopropylphenyl. |
 |
MIC value of 0.5 μg/mL against M. tuberculosis (H37Ra). |
[110] |
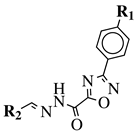 |
R1 = H, F, Cl, Br, methyl, nitro, methoxy or hydroxy;
R2 = 4-hydroksy-3-methoxyphenyl, 2-styryl, ferrocene or 5-benzo[1,3]dioxole. |
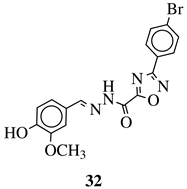 |
IC50 value of 0.02 μM against P. falciparum.
In vivo studies failed—none in vivo activity. |
[111] |
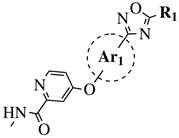 |
R1 = Me, Et, cyclopropyl, iPr, CF3, iBu or CH2OCH3;
Ar1 = p-phenylene, p-2-methylphenylene, p-2,6-dimethylphenylene, 2,5-pyridinyl or 3-methylbenzothiophene |
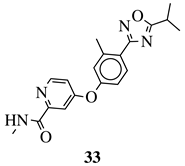 |
IC50 values of 66.0, 22.0 and 3.7 nM against hRV-B14, hRV-A21 and hRV-A71, respectively. |
[112] |