Hereditary angioedema is a rare inherited disorder characterized by recurrent episodes of the accumulation of fluids outside of the blood vessels, causing rapid swelling of tissues in the hands, feet, limbs, face, intestinal tract, or airway.
- HAE
- C1-INH-HAE
- nC1-INH-HAE
- SERPING1
- F12
- PLG
1. Introduction
Angioedema is characterized by a localized, self-limiting, and transient subcutaneous or submucosal swelling, which can present with or without episodes of urticaria, and usually subsides within 24–37 h. The clinical expression is highly variable, from asymptomatic individuals to patients suffering from disabling and life-threatening attacks with a demonstrated humanistic and economic burden [1]. Manifestations may imply swelling of the extremities and superficial regions of the face; affect the gastrointestinal tract, because of edema of the bowel wall, and cause severe cramping abdominal pain, nausea, vomiting and, sometimes, diarrhea; and occasionally compromise breathing, including laryngeal edema and/or severe tongue/pharyngeal edema so that secretions cannot be handled [2]. The severity and frequency of acute attacks of angioedema are variable, ranging from once/year to three attacks/week [3]. Although a series of predisposing circumstances, including trauma, fluctuating hormone level (particularly increased estrogen), infection, and severe emotional stress have been identified, most acute episodes of angioedema seem apparently spontaneous. In the general population, an acute episode of angioedema has been estimated to occur in up to 7.4% of subjects during their lifetime [4]. Thus, after excluding cases of angioedema with identifiable cause or wheals, one person in a hundred is likely to have an episode of angioedema. Approximately 0.05% of them have been estimated to suffer from recurrent/nonallergic angioedema [5].
2. Hereditary Angioedema
Hereditary angioedema (HAE) is a genetic disorder that predisposes an individual to develop vasogenic edema. Prevalence of HAE has been reported to be 1 in 10,000 to 1 in 150,000 [6]. HAE shows no ethnic- or sex-based differences but tends to be more severe in women [2][7]. The pathogenesis of HAE involves the accumulation of extravascular fluid in various tissues via a non-inflammatory and non-allergic mechanism. Clinical manifestations include abrupt onset swelling around the eyes, face and extremities; pain in theabdomen (as a result of bowel edema), and laryngeal edema leading to hoarseness of voice, breathing difficulty, and occasionally death [8][9].
Several disorders may manifest with subcutaneous or submucosal swelling. The presence of severe swelling can be mistaken for an allergic reaction or acute abdominal condition. Misdiagnosis can lead to ineffective therapies and unnecessary surgeries [10]. In 1963, Donaldson and Evans discovered that HAE was caused by a genetic deficiency of the C1 inhibitor (C1-INH) [11]. Since then, many large studies have established that the most common type of hereditary angioedema (HAE) is the result of gene mutations resulting in reduced C1-INH functional plasma. This results in two HAE variants of C1-INH-HAE: type 1 and type 2. C1-INH type 1 results from a failure to synthesize C1-INH, whereas in type 2, an abnormal, non-functional protein is synthesized.
The increase in vascular permeability that causes angioedema in HAE is related to the mediators of the contact system or kallikrein–kinin pathway. C1-INH regulates the contact system through the inhibition of plasma kallikrein and coagulation factor FXIIa. The loss of the inhibitory activity of C1-INH leads to bradykinin overproduction. It has been documented that an increased release of bradykinin is the cause of angioedema via its action on B2 receptors leading to an increase in vascular permeability [12][13]. Moreover, a dominantly inherited disease has been described that has a similar clinical picture to C1-INH-HAE, but with normal C1-INH level and activity. This new type of HAE has no mutation in the SERPING1 gene and it is classified as nC1-INH-HAE (Figure 1).
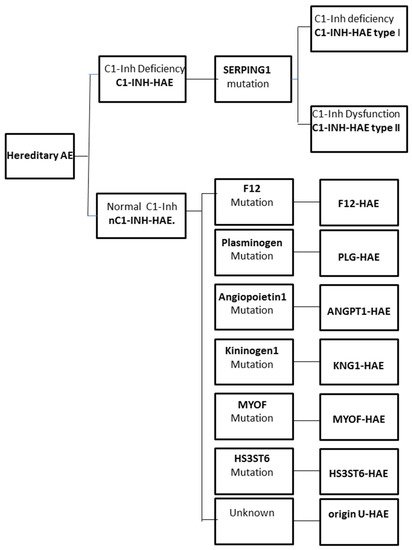
Figure 1. Classification of the different forms of hereditary angioedema (HAE).
2.1. Genetics of C1-INH-HAE
C1-INH-HAE is the most frequent type of HAE due to low production or nonfunctional serine protease inhibitor, namely C1 esterase inhibitor (C1-INH). The autosomal-dominant mode of inheritance has been described for C1-INH-HAE when one of the two alleles of the C1-INH gene (SERPING1) is mutated [14][15][16].
Mutations in SERPING1, the gene that encodes for C1-INH, are responsible for the majority of cases of hereditary angioedema. Mutations in the SERPING1 gene may be classified as shown in Figure 2.
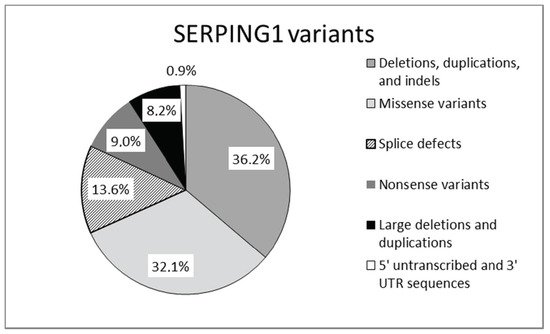
Figure 2. Mutations reported in SERPING1 gene [17].
Mutations leading to type 1 are dispersed throughout the entire SERPING1. Type 2 mutations map around the protein reactive center loop (RCL), with the single exception of a mutation in the amino acid residue Lys251, which affects functionality after protein folding [15][16]. This gene is characterized by a great allelic heterogeneity and approximately 748 different mutations have been reported and collected in the Human Gene Mutation Database (HGMD) and in a specific, online HAE database (HAEdb, hae.enzim.hu) [17][18][19]. The vast majority were identified as heterozygous variants (729), though homozygous variants in 10 probands and compound heterozygous variants (nine combinations) were found. In addition, probands were identified as de novo cases and six probands with heterozygous variants exhibited gonadal mosaicism [17].
The severity and course of HAE may vary greatly even among family members harboring the same mutation. A correlation between different types of mutations and clinical phenotype is controversial. Only patients carrying missense mutations leading to the change of a single amino acid exhibited a less severe clinical phenotype [20]. The involvement of epigenetic changes [21] and environmental factors (i.e., temperature, pH, and oxidative stress) in the pathogenesis of HAE, has also been postulated [16].
Recently the advent of new technology allowed the identification of novel SERPING1 variants in C1-INH-HAE patients in particular pathogenic intronic regions. These are hardly detectable with a direct sequencing approach. Using NGS technology two pathogenic deep intronic variants have been detected: the c.-22- 155G > T located at intron 1 and the c.1029 + 384A > G located at intron 6 [22][23][24].
2.2. The Genetics of nC1-INH-HAE
In 2000, separately Bork et al. [25] and Binkley et al. [26] described another type of HAE with a quantitatively and qualitatively normal C1-INH (labeled as nC1-INH-HAE). In this rare type of HAE, clinical manifestations were similar to C1INH deficiency HAE, except that there is a higher female predominance due to aggravation during pregnancy and estrogen dependency. In these groups of patients no mutation was detected in the SERPING1 gene. In 2006 two missense mutations (p.Thr328Lys and p.Thr328Arg) in the F12 gene were detected for the first time in six out of twenty German families with nC1-INH-HAE [27]. Other rare pathogenic mutations in this gene have been identified at the same locus in different cohorts: F12 gene encoding for the coagulation factor XII. HAE-FXII is inherited as an autosomal dominant trait with incomplete penetrance and is the result of a gain-of-function mutation in the F12 gene. All of these mutations occur at exon 9 of F12 which encodes a highly glycosylated region of the protein and leads to an increased production of activated Factor XII (Factor XIIa) via activation by plasmin [27][28][29][30][31][32].
Table 1 summarizes the mutations described in F12 and in the other genes associated with nC1-INH-HAE.
Table 1. Summary of the mutations in nC1-INH-HAE.
| F12 | T328K T328R c.971_1018 + 24del72 c.892_909dup |
| PLG | K330E |
| ANGPT1 | A119S A8V Q370H |
| KNG1 | M379K |
| MYOF | R217S |
| HS3ST6 | T144S |
In the following years families or individuals with nC1-INH-HAE, in particular families with nC1-INH-HAE and without mutation in the F12 gene, were analyzed often by whole-exome sequencing (WES), but also by a direct candidate gene approach [33][34][35][36][37][38][39]. These studies allowed the identification of five new genes linked nC1-INH-HAE. In 2018 a novel variant in the plasminogen gene (PLG) was described in patients with nC1-INH-HAE [33][34]. The mutation c.988A > G, located in exon 9 of the PLG gene, leads to the missense mutation p.Lys330Glu (K330E) in the kringle 3 domain of the PLG protein. In large multiple-case families, co-segregation of the mutation with the disease phenotype was demonstrated; the inheritance pattern was autosomal dominant, with incomplete penetrance [33][34]. This mutation has been reported in more than 14 patients with nC1-INH-HAE belonging to 4 families and was transmitted as an autosomal dominant trait presenting incomplete penetrance [33]. This mutation has been reported in more than 125 affected individuals from different countries [40]. Plasminogen is the inactive precursor protein of the enzyme plasmin. Plasmin plays a role in bradykinin production via activation of factor XII. The mutant protein leads to the increased production of the bradykinin.
In the same year, in affected female members of a large nC1-INH-HAE family, a missense variant in the angiopoietin-1 gene (ANGPT1), c.355G > T (p.Ala119Ser), was identified [35]. This variation disrupts the multimerization of the protein and affects the protein’s abilities to bind its specific receptor on endothelial cells suggesting a novel and independent mechanism leading to vascular permeability and angioedema through a mechanism of haploinsufficiency [41]. Other potentially pathogenic variants of ANGPT1 (p.Ala8Val; p.Gln370His) were found by Cagini et al. [42].
Recently, WES has led to the identification of another two causal variants in two nC1-INH-HAE families. Bork et al. [36] identified a novel variant of the KNG1 gene encoding the high molecular weight as well as low molecular weight kininogen (proteins). The novel mutation (p.Met379Lys) resulted in the substitution of a methionine with lysine at position 379 and co-segregated with clinical symptoms of hereditary angioedema (HAE) with normal C1-INH levels in three generations of a large German family.
In 2020 in an Italian family, a rare Myoferlin variant (MYOF Arg217Ser) was identified and three out of four carriers were symptomatic. The MYOF-217S variant acts with a gain of function mechanism and allows a higher ability of the mutant protein to localize VEGFR-2 to the plasma membrane in response to VEGF stimuli, which suggests the involvement of VEGF-mediated signaling in HAE. The identification of the MYOF-217S variant as causative of HAE suggests an involvement of the VEGF-mediated signaling in HAE pathophysiology [38].
By performing whole exome sequencing on a multigenerational family with nC1-INH-HAE, a mutation in the heparan sulfate (HS)-glucosamine 3-O-sulfotransferase 6 (HS3ST6) has been identified. This mutation, c.430A > T (p.Thr144Ser), has been found in all three affected family members. This gene encodes HS-glucosamine 3-O-sulfotransferase 6 (3-OST-6), which is involved in the last step of HS biosynthesis. The p.Thr144Ser mutation is hypothesized to result in incomplete HS biosynthesis and is likely to affect cell surface interactions of key players in angioedema formation and represent a novel mechanism for disease development [39].
2.3. Disease-Modifying Factors
The high variability in clinical expression between patients with the same mutation led to the hypothesis that mutations in these and other related genes could be potential modifiers in the clinical phenotype of patients with known genetic causes of HAE. Several polymorphisms in different genes and their effects on the clinical phenotype of patients were analyzed. These included p.Y244C, p.G354R, and p.T916M in the ACE gene; p.C548Y in the KLKB1 (kallikrein) gene; and p.D287N in the NOS3 (nitric oxide synthase) gene [43]. However, the role of these polymorphisms on modification in the clinical phenotype of HAE remains to be clarified. Other disease-modifying factors, F12 or KLKB1 gene polymorphisms, have been studied to explain clinical variability of C1-INH-HAE or nC1-INH-HAE. The possible associations of the functional promoter F12-46C/T polymorphism (rs1801020) with clinical features of C1-INH-HAE and the SERPING1 mutational status have been investigated. The F12-46C/T carriage acts as an independent modifier of C1-INH-HAE severity regardless of SERPING1 mutational status [44][45]. The c.-4C/T polymorphism (rs1801020) in the 5-UTR region of the F12 gene was shown to significantly influence the degree of contact system activation and the clinical severity of the disease. Patients carrying FXII-HAE (p.Thr309Lys variant) and c.-46CC genotype exhibited more severe and frequent manifestations of the disease [46].
The c.428G/A (rs3733402) polymorphism in the KLKB1 gene, encoding plasma kallikrein, was also investigated. Carriers of the G allele of the KLKB1-428G/A polymorphism exhibited a significantly delayed disease onset [47]. Since an earlier onset of symptoms is inversely correlated with the subsequent course of the disease, these polymorphisms may be helpful prognostic biomarkers of disease severity.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10092023
References
- Cicardi, M.; Aberer, W.; Banerji, A.; Bas, M.; Bernstein, J.A.; Bork, K.; Caballero, T.; Farkas, H.; Grumach, A.; Kaplan, A.P.; et al. Classification, diagnosis, and approach to treatment for angioedema: Consensus report from the Hereditary Angioedema International Working Group. Allergy 2014, 69, 602–616.
- Zuraw, B.L. Hereditary Angioedema. N. Engl. J. Med. 2008, 359, 1027–1036.
- Bork, K.; Meng, G.; Staubach, P.; Hardt, J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion 2005, 45, 1774–1784.
- Madsen, F.; Attermann, J.; Linneberg, A. Epidemiology of Non-hereditary Angioedema. Acta Derm. Venereol. 2012, 92, 475–479.
- Kaplan, A.P. Angioedema. World Allergy Organ. J. 2008, 1, 103–113.
- Ghazi, A.; Grant, J.A. Hereditary angioedema: Epidemiology, management, and role of icatibant. Biologics 2013, 7, 103–113.
- Nzeako, U.C.; Frigas, E.; Tremaine, W.J. Hereditary angioedema: A broad review for clinicians. Arch. Intern. Med. 2001, 161, 2417–2429.
- Jalaj, S.; Scolapio, J.S. Gastrointestinal manifestations, diagnosis, and management of hereditary angioedema. J. Clin. Gastro-enterol. 2013, 47, 817–823.
- Xu, Y.-Y.; Zhi, Y.-X.; Liu, R.-L.; Craig, T.; Zhang, H.-Y. Upper airway edema in 43 patients with hereditary angioedema. Ann. Allergy Asthma Immunol. 2014, 112, 539–544.e1.
- Zanichelli, A.; Longhurst, H.J.; Maurer, M.; Bouillet, L.; Aberer, W.; Fabien, V.; Andresen, I.; Caballero, T.; Aberer, W.; Grumach, A.; et al. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy Asthma Immunol. 2016, 117, 394–398.
- Donaldson, V.H.; Evans, R.R. A biochemical abnormality in hereditary angioneurotic edema: Absence of serum inhibitor of C’1-esterase. Am. J. Med. 1963, 35, 37–44.
- Kaplan, A.P.; Joseph, K. Pathogenic mechanisms of bradykinin mediated diseases: Dysregulation of an innate inflammatory pathway. Adv. Immunol. 2014, 121, 41–89.
- Maurer, M.; Bader, M.; Bas, M.; Bossi, F.; Cicardi, M.; Cugno, M.; Howarth, P.; Kaplan, A.; Kojda, G.; Leeb-Lundberg, F.; et al. New topics in bradykinin research. Allergy 2011, 66, 1397–1406.
- Banday, A.Z.; Kaur, A.; Jindal, A.K.; Rawat, A. Singh S An update on the genetics and pathogenesis of hereditary angioedema. Genes Dis. 2020, 7, 75–83.
- Zuraw, B.L. Hereditary angioedema with normal C1 inhibitor: Four types and counting. J. Allergy Clin. Immunol. 2018, 141, 884–885.
- Caccia, S.; Suffritti, C.; Carzaniga, T.; Berardelli, R.; Berra, S.; Martorana, V.; Fra, A.; Drouet, C.; Cicardi, M. Intermittent C1-inhibitordeficiency associated with recessive inheritance: Functional and structural insight. Sci Rep. 2018, 8, 977.
- Ponard, D.; Gaboriaud, C.; Charignon, D.; Ghannam, A.; Wagenaar-Bos, I.G.A.; Roem, D.; López-Lera, A.; López-Trascasa, M.; Tosi, M.; Drouet, C. SERPING1 mutation update: Mutation spectrum and C1 Inhibitor phenotypes. Human Mutat. 2020, 41, 38–57.
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Qual. Life Res. 2014, 133, 1–9.
- Kalmár, L.; Hegedüs, T.; Farkas, H.; Nagy, M.; Tordai, A. HAEdb: A novel interactive, locus-specific mutation database for the C1 inhibitor gene. Human Mutat. 2005, 25, 1–5.
- Bors, A.; Csuka, D.; Varga, L.; Farkas, H.; Tordai, A.; Fust, G.; Szilagyi, A. Less severe clinical manifestations in patients with hereditary angioedema with missense C1INH gene mutations. J. Allergy Clin. Immunol. 2003, 131, 1708–1711.
- Khan, S.; Longhurst, H. Epigenetic alterations on C1-inhibitor expression may influence hereditary angioedema attack fre-quency and C4 levels. Clin. Exp. Immunol. 2020, 202, 144–145.
- Vatsiou, S.; Zamanakou, M.; Loules, G.; Psarros, F.; Parsopoulou, F.; Csuka, D.; Valerieva, A.; Staevska, M.; Porebski, G.; Obtulowicz, K.; et al. A novel deep intronic SERPING1 variantas a cause of hereditary angioedema due to C1-inhibitor deficiency. Allergol Int. 2020, 69, 443–449.
- Hujová, P.; Souček, P.; Grodecká, L.; Grombiříková, H.; Ravčuková, B.; Kuklínek, P.; Hakl, R.; Litzman, J.; Freiberger, T. Deep intronic mutation inSERPING1 caused hereditary angioedema through pseudoexon activation. J. Clin. Immunol 2020, 40, 435–446.
- Loules, G.; Zamanakou, M.; Parsopoulou, F.; Vatsiou, S.; Psarros, F.; Csuka, D.; Porebski, G.; Obtulowicz, K.; Valerieva, A.; Staevska, M.; et al. Targeted next-generation sequencing for the molecular diagnosis of hereditary angioedema due to C1-inhibitor defi-ciency. Gene 2018, 667, 76–82.
- Bork, K.; Barnstedt, S.E.; Koch, P.; Traupe, H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet 2000, 356, 213–217.
- Binkley, K.E.; Davis, A. Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioedema. J. Allergy Clin. Immunol. 2000, 106, 546–550.
- Dewald, G.; Bork, K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 2006, 343, 1286–1289.
- Bork, K.; Gül, D.; Hardt, J.; Dewald, G. Hereditary Angioedema with Normal C1 Inhibitor: Clinical Symptoms and Course. Am. J. Med. 2007, 120, 987–992.
- Kiss, N.; Barabás, E.; Várnai, K.; Halász, A.; Varga, L.Á.; Prohászka, Z.; Farkas, H.; Szilágyi, Á. Novel duplication in the F12 gene in a patient with recurrent angioedema. Clin. Immunol. 2013, 149, 142–145.
- Bork, K.; Wulff, K.; Hardt, J.; Witzke, G.; Lohse, P. Characterization of a partial exon 9/intron 9 deletion in the coagulation factor XII gene (F12) detected in two Turkish families with hereditary angioedema and normal C1 inhibitor. Haemophilia 2014, 20.
- Bork, K.; Wulff, K.; Meinke, P.; Wagner, N.; Hardt, J.; Witzke, G. A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1-inhibitor. Clin. Immunol. 2011, 141, 31–35.
- De Maat, S.; Björkqvist, J.; Suffritti, C.; Wiesenekker, C.P.; Nagtegaal, W.; Koekman, A.; van Dooremalen, S.; Pasterkamp, G.; de Groot, P.G.; Cicardi, M.; et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J. Allergy Clin. Immunol. 2016, 138, 1414–1423.e9.
- Bork, K.; Wulff, K.; Steinmüller-Magin, L.; Braenne, I.; Staubach-Renz, P.; Witzke, G.; Hardt, J. Hereditary angioedema with a mutation in the plasminogen gene. Allergy 2018, 73, 442–450.
- Dewald, G. A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angi-oedema with normal C1 inhibitor. Biochem Biophys Res. Commun. 2018, 498, 193–198.
- Bafunno, V.; Firinu, D.; D’Apolito, M.; Cordisco, G.; Loffredo, S.; Leccese, A.; Bova, M.; Barca, M.P.; Santacroce, R.; Cicardi, M.; et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J. Allergy Clin. Immunol. 2018, 141, 1009–1017.
- Bork, K.; Wulff, K.; Rossmann, H.; Steinmüller-Magin, L.; Braenne, I.; Witzke, G.; Hardt, J. Hereditary angioedema cosegre-gating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy 2019, 74, 2479–2481.
- Loules, G.; Parsopoulou, F.; Zamanakou, M.; Csuka, D.; Bova, M.; González-Quevedo, T.; Psarros, F.; Porebski, G.; Speletas, M.; Firinu, D.; et al. Deciphering the Genetics of Primary Angioedema with Normal Levels of C1 Inhibitor. J. Clin. Med. 2020, 9, 3402.
- Ariano, A.; D’Apolito, M.; Bova, M.; Bellanti, F.; Loffredo, S.; D’Andrea, G.; Intrieri, M.; Petraroli, A.; Maffione, A.B.; Spadaro, G.; et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy 2020, 75, 2989–2992.
- Bork, K.; Wulff, K.; Möhl, B.S.; Steinmüller-Magin, L.; Witzke, G.; Hardt, J.; Meinke, P. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J. Allergy Clin. Immunol. 2021, 0091.
- Recke, A.; Massalme, E.G.; Jappe, U.; Steinmüller-Magin, L.; Schmidt, J.; Hellenbroich, Y.; Hüning, I.; Gillessen-Kaesbach, G.; Zillikens, D.; Hartmann, K. Identification of the recently described plasminogen gene mutation p.Lys330Glu in a family from Northern Germany with hereditary angioedema. Clin. Transl. Allergy 2019, 9, 1–4.
- D’Apolito, M.; Santacroce, R.; Colia, A.L.; Cordisco, G.; Maffione, A.B.; Margaglione, M. Angiopoietin-1 haploinsufficiency affects the endothelial barrier and causes hereditary angioedema. Clin. Exp. Allergy 2019, 49, 626–635.
- Cagini, N.; Veronez, C.L.; Azevedo, B.F.; Constantino-Silva, R.N.; Martin, R.P.; Da Silva, J.; Grumach, A.S.; Pesquero, J.B. In silico Analysis Of Alterations In ANGPT1 Gene Supports A New Pathway Responsible To Mediate Hereditary Angioedema In Brazilian Patients with No Mutations in SERPING1 and F12 Genes. J. Allergy Clin. Immunol. 2018, 141, AB46.
- Veronez, C.L.; Aabom, A.; Martin, R.P.; Filippelli-Silva, R.; Gonçalves, R.F.; Nicolicht, P.; Mendes, A.R.; Da Silva, J.; Guilarte, M.; Grumach, A.S.; et al. Genetic Variation of Kallikrein-Kinin System and Related Genes in Patients with Hereditary Angioedema. Front. Med. 2019, 6, 6–28.
- Speletas, M.; Szilágyi, Á.; Csuka, D.; Koutsostathis, N.; Psarros, F.; Moldovan, D.; Magerl, M.; Kompoti, M.; Varga, L.; Maurer, M.; et al. F12-46C/T polymorphism as modifier of the clinical phenotype of hereditary angioedema. Allergy 2015, 70, 1661–1664.
- Grivčeva-Panovska, V.; Košnik, M.; Korošec, P.; Andrejević, S.; Karadža-Lapić, L.; Rijavec, M. Hereditary angioedema due to C1-inhibitor deficiency in Macedonia: Clinical characteristics, novel SERPING1 mutations and genetic factors modifying the clinical phenotype. Ann. Med. 2018, 50, 269–276.
- Corvillo, F.; de la Morena Barrio, M.E.; Marcos Bravo, C.; López-Trascasa, M.; Vicente, V.; Emsley, J.; Caballero, T.; Corral, J.; Lopez Lera, A. The FXII c.-4T>C Polymorphism as a Disease Modifier in Patients with Hereditary Angioedema Due to the FXII p.Thr328Lys Variant. Front. Genet. 2020, 11, 1033.
- Gianni, P.; Loules, G.; Zamanakou, M.; Kompoti, M.; Csuka, D.; Psarros, F.; Magerl, M.; Moldovan, D.; Maurer, M.; Speletas, M.G.; et al. Genetic Determinants of C1 Inhibitor Deficiency Angioedema Age of Onset. Int. Arch. Allergy Immunol. 2017, 174, 200–204.