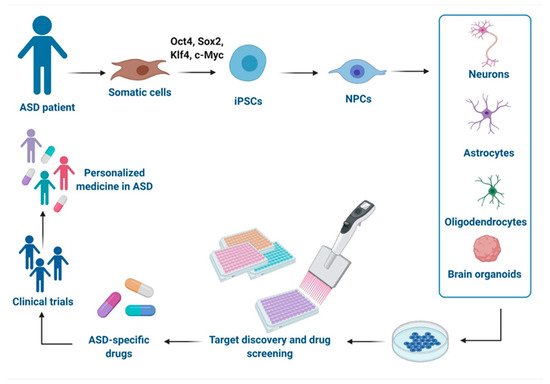
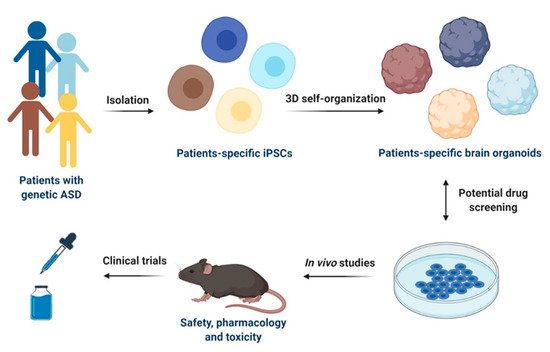
2.1. iPSCs-Derived Neural Progenitor Cells
NPCs are a population of cells with self-renewal and multipotent differentiation ability that can differentiate into both mature neurons and glial cells. NPCs play an important role in neurogenesis [
34], and their dysfunction has been related with both non-syndromic [
35,
36,
37,
38] and syndromic ASD, including RETT syndrome (RTT) [
39,
40,
41,
42], Fragile X syndrome (FXS) [
43,
44], tuberous sclerosis complex (TSC) [
45,
46], Phelan-McDermid syndrome (PMDS) [
47], and Timothy syndrome (TS) [
48,
49].
In regard to idiopathic ASD, NPCs from individuals with macrocephaly displayed an increased cellular proliferation resulted from alterations in a canonical Wnt-β-catenin/BRN transcriptional cascade [
35]. These abnormalities in proliferation lead to aberrant neurogenesis and reduced synaptogenesis, thus contributing to functional defects in neuronal networks [
35]. Consistently, a recent study demonstrated that the hyperproliferation observed in NPCs derived from iPSCs of ASD subjects with macrocephaly affects genome instability by inducing replication stress-associated genes [
36]. Other authors reported that iPSCs-derived NPCs show an impaired crosstalk between the pathways of mTORC1 and Reelin-DAB1, which is known to play a crucial role in regulating neuronal migration and synapse function [
37]. This altered interplay was already shown to affect neuronal migration and positioning in a TSC mouse model and in cortical tubers from TSC patients [
50]. Moreover, abnormalities in neuronal development, morphology and function were observed in NPCs from a non-syndromic ASD child with a de novo balanced translocation disrupting
TRPC6, encoding for the human transient receptor potential 6 channel, involved in excitatory synapse and dendritic spine formation [
38].
NPCs were also obtained from patients affected by RTT (OMIM #312750), an early-onset neurodevelopmental disorder caused by mutations in the X-linked gene methyl-CpG-binding protein 2 (
MECP2; Xp28), a ubiquitously expressed transcriptional regulator essential for the maturation and normal function of neurons [
51,
52]. This syndrome is the second most common cause of severe intellectual disability in females and, a considerable fraction of affected individuals meets diagnostic criteria for ASD [
53]. Atypical RTT variants have been identified, associated with mutations in cyclin-dependent kinase-like 5 (
CDKL5; Xp22.13) and forkhead box protein G1 (
FOXG1; 14q12) [
51]. NPCs derived from iPSCs of patients carrying different
MECP2 mutations underwent X-inactivation when differentiating into functional neurons [
39]. Another study found an increase in neural long-interspersed nuclear element-1 (LINE-1) retrotransposon, suggesting that MECP2 is involved in its mobility within the CNS [
40]. In three children with
MECP2 duplication, an altered expression of neuronal progenitor genes in NPCs was found [
41]. Interestingly, a recent report demonstrated that NPCs and also cortical neurons show a global repressed translation and a decreased ribosome engagement of NEDD4-family ubiquitin ligases, leading to accumulation of target proteins that escape proteasome degradation. These evidences provide insight into novel therapeutic strategies based on the regulation of ubiquination process [
42].
FXS (OMIM #300624) is responsible for the most common monogenic cause of ASD that is typically due to a triplet repeat expansion (>200 CGG repeats in the 5′ untranslated region) and subsequent methylation of the
FMR1 gene on the X chromosome (Xq27.3) [
54]. As a result, the amount of FMRP (protein product of
FMR1) decreases, leading to an abnormal translation of different proteins. In fact, FMRP is an mRNA binding protein that regulates the mRNA transport and translational regulation in dendrites [
54]. In the case of FXS, Telias et al. performed directed differentiation of cells from three patients and observed in FXS neural progenitors an abnormal expression of key NPC genes (
SOX1,
NOTCH1,
PAX6) [
43]. The iPSCs-derived FXS NPCs were then used to investigate functional maturation of the excitatory transmission system in response to the glutamate analog (AMPA) [
44]. The authors demonstrated an impairment in calcium (Ca
2+) signaling via AMPA receptors (AMPARs), suggesting that their functional alterations affect neuronal differentiation and contribute to aberrant neuronal circuit formation and function in FXS [
44].
Another ASD-associated syndrome where iPSCs were obtained and studied is TSC (OMIM #191100 and #613254), an autosomal dominant genetic disorder, characterized by benign tumors in the brain and other organs, epilepsy, cognitive impairment, and high penetrance of ASD [
55]. The prevalence of ASD in TSC varies among studies, but it is estimated to range from 36% to 50% [
55]. TSC is caused by mutations in two genes:
TSC1 (hamartin gene, 9q34) and
TSC2 (tuberin gene, 16p13.3) that act as tumor suppressors inhibiting the mammalian target of rapamycin (mTOR) [
56]. These events lead to constitutive activation of mTOR complex 1 cascade and the uncontrolled proliferation of cells. In neurons, dysfunction of this pathway results in abnormal development of fundamental processes that have been proposed to contribute to the behavioral deficits seen in ASD [
55]. Human iPSCs-derived from TSC patients exhibited a delay in their ability to differentiate into neurons that is probably related to a dysregulated PI3K/AKT signaling [
45]. Recently, isogenic NPCs derived from a patient carrying a nonsense
TSC1 mutation revealed altered early neurodevelopmental phenotypes displaying enhanced proliferation, aberrant neurite outgrowth and enlarged cell size, consistent with mTORC1 activation [
46]. However, rapamycin treatment was effective only in reverting enlarged cell size, whereas it was ineffective in increasing proliferation and neurite outgrowth in iPSCs-derived NPCs. Thus, it could be speculated that early neurodevelopmental phenotypes due to
TSC1 loss are not solely related to mTORC1 activation. Moreover, transcriptome analysis showed differentially expressed genes (DEGs) related to ASD phenotype with a genotype-dependent linear response: upregulated/downregulated genes were further increased/decreased in homozygous NPCs, generated by CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 nuclease) genome editing technology, compared to the heterozygous state [
46].
Similarly, genome-wide RNA sequencing was applied to explore DEGs in iPSCs-NPCs and forebrain neurons derived from PMDS probands and unaffected siblings [
47]. PMDS (OMIM #606232), also called 22q13 deletion syndrome, is a rare disease with different clinical features, such as severe intellectual impairments and ASD. A critical region involved three genes, but
SHANK3 is the strongest candidate for the neurobehavioral symptoms [
57]. In fact,
SHANK3 encodes a scaffolding protein enriched in the postsynaptic density of glutamatergic synapses and plays a critical role in synaptic function and dendrite formation [
58]. In PMDS-iPSCs, the authors found an underexpression of genes involved in Wnt signaling, embryonic development, and protein translation, whereas an overexpression was observed in genes related to pre- and post-synaptic signaling, synaptic plasticity regulation and G-protein-gated potassium channels. Interestingly, these data partially overlapped with iPSC transcriptome findings in other ASDs, converging on altered Wnt signaling and extracellular matrix [
47].
In 2011, patient-specific iPSCs-derived NPCs and cortical neurons offered new insights into the pathogenesis of TC (OMIM #601005), a rare and lethal multi-organ disorder connected with gain-of-function missense mutation in the
CACNA1C gene (12p13.33) [
59], linking its phenotype to defects in Ca
2+ signaling and gene expression-dependent activity [
48]. In the same study, the authors reported an abnormal expression of tyrosine hydroxylase (TH) and an increased production of norepinephrine and dopamine in neurons, consistent with the key role of catecholamines in sensory gating and social behavior [
48]. A genome-wide weighted co-expression network analysis (WGCNA) on NPCs from TS-iPSCs further confirmed dysregulation of Ca
2+ signaling in neural development and function [
49].
2.2. iPSCs-Derived Neurons
Neurons, generated from NSCs during brain development, are electrically excitable cells that transmit information via specialized connections called synapses [
60]. Post-mortem human brain analysis, as well as functional and structural imaging studies in ASD, showed an impairment in the formation of neuronal networks and synaptogenesis [
61,
62]. In addition, cerebral tissues displayed altered neuronal phenotype, consisting of reduced soma size, abnormal neuronal morphology, reduced dendritic arborization, fewer dendritic spines and synapses [
63,
64]. Similar morphological alterations were also found in iPSCs-derived neurons from non-syndromic ASD patient carrying a
TRPC6 mutation [
38]. The same authors demonstrated that the treatment with insulin-like growth factor 1 (IGF-1) or hyperforin, a TRPC6-specific agonist, is able to rescue the neuronal abnormalities, suggesting a possible target for therapeutic strategies in individuals with alterations in this pathway. Interestingly, it has been also shown that MECP2 levels affect
TRPC6 expression, hypothesizing common pathways among ASD [
38]. Additional iPSCs-based studies in idiopathic ASD reported an imbalance between excitatory and inhibitory synapses, leading to deficits in social interaction and behaviors as widely described in autistic individuals [
65,
66,
67,
68]. An overproduction of GABAergic inhibitory neurons has been observed in iPSCs-derived neurons from patients carrying
FOXG1 mutations [
69]. In ASD individuals with macrocephaly, other authors reported a reduced synaptogenesis, abnormal neurogenesis and a significant decrease in both inhibitory and excitatory neurotransmitters, leading to functional defects in neuronal networks which were rescued by IGF-1 treatment [
45]. A reduced activity of neurons was further observed in a cohort of non-syndromic ASD patients, showing a significant decrease in excitatory neurotransmitter release, synaptic events and, consequently, spontaneous spike rate [
31]. In iPSCs-derived neurons from non-syndromic individuals carrying
SHANK2 mutations, other authors recently showed a hyperconnectivity as demonstrated by an increase in dendrite length and complexity, synapse number and frequency of spontaneous excitatory post-synaptic currents [
70].
Human iPSCs-derived neurons have been largely used to study a wide range of syndromic ASD forms. Concerning RTT, several groups generated iPSC lines from patients and successfully differentiated them into NPCs and functional neurons. Similarly to results reported for NPCs, iPSCs-derived neurons exhibited increased susceptibility for LINE-1 retrotransposition, probably caused by
MECP2 mutations [
40]. Marchetto and collaborators [
39] showed smaller soma size, reduced dendritic spine densities and fewer synapses in RTT iPSCs-derived neurons in comparison with the isogenic wild-type ones. Moreover, they observed alterations in Ca
2+ influx, thus causing electrophysiological defects in the RTT neurons [
39]. Conversely, other authors reported that cortical neurons, derived from iPSC lines with
MECP2 duplicated gene, display increased synaptogenesis and dendritic complexity with altered neuronal network synchronization recorded by multi-electrode array (MEA) electrophysiology technique [
41]. The functional phenotype was further rescued by the treatment with one histone deacetylase inhibitor, named NCH-51 [
51]. RNA-seq profiling on differentiated iPSCs-neurons from RTT patients harboring different
MECP2 mutations revealed a prominent enrichment in GABA pathway genes, including GABA receptors and other GABA circuits [
71]. Intriguingly,
MECP2-mutated neurons showed an impairment of the microtubule network along with a significant decrease of acetylated α-tubulin which was reverted by treatment with selective inhibitors of histone deacetylase 6 (HDAC6), the main cytoplasmic deacetylase in which the main substrate is acetylated α-tubulin [
71]. In regard to CDKL5 deficiency disorder (CDD), an atypical form of RTT, a study successfully generated clones of
CDKL5-mutated iPSCs to model disease pathogenesis in vitro [
72]. CDD is an X-linked neurological disease caused by pathogenic mutations in the gene for cyclin-dependent kinase-like 5 (
CDKL5), a serine-threonine kinase highly expressed in the brain [
73]. This rare disease was considered as an atypical variant of RTT; however, it has since been recognized as a distinct disorder with common clinical features, such as early-life seizures, autistic behaviors, and intellectual disability [
74]. Interestingly,
CDKL5-mutated iPSCs from females maintained X-chromosome inactivation, allowing the use of clones expressing the wild-type allele as ideal experimental controls, genetically identical to those derived from the same patient [
72]. In iPSC-derived neurons from patients carrying
CDKL5 mutations, a following study detected a loss of synaptic contacts, as well as an increased number of aberrant dendritic spines, demonstrating an interesting role for
CDKL5 in spine development and synapse morphogenesis [
75].
The majority of studies performed in iPSCs-derived neurons from FXS patients revealed an impairment of neuronal differentiation and maturation as a result of epigenetic differences on
FMR1 gene expression [
76,
77,
78,
79,
80]. Moreover, neurons exhibited a number of additional phenotypic abnormalities, including an altered electrophysiological network activity, neurite outgrowth and branching defects [
80,
81]. Perturbations in synaptic transmission, cell proliferation and ion transmembrane transporter activity pathways were also reported [
80]. Importantly, other authors demonstrated dysregulated Ca
2+ signals in iPSCs-derived neurons, strengthening the key role of intracellular Ca
2+ in neurite growth and synaptic connections [
82].
In TSC, iPSCs-derived neurons presented with an enlarged soma, decreased neurite length, complex neurite branching and abnormal connections among cells, as compared to those from unaffected individuals [
83]. These abnormalities may be related to hyperactivity of mTOR. Similarly, Zucco et al. generated iPSC-derived NPCs and neurons from two TSC patients reporting a dysregulation of PI3K/AKT/mTORC1 pathway [
45]. In mono-cultures of iPSCs-derived cortical neurons, MEA analysis showed an increase in basal dendritic branching and spontaneous Ca
2+ event frequency [
84], consistent with the network hyperactivity previously observed in the TSC mouse model [
85]. It should be noted that these aforementioned studies were performed in iPSCs-derived neurons from TSC patients with heterozygous mutations. In a recent study examining iPSCs-derived neurons carrying either single or biallelic mutations in
TSC2, the authors reported that the loss of one or both alleles of
TSC2 results in mTORC1 hyperactivation and specific neuronal abnormalities which were partially rescued by pharmacological treatment with mTOR regulator rapamycin [
86]. However, only neurons harboring biallelic mutations displayed hyperactivity and upregulation of cell adhesion genes observed in cortical tubers. Collectively, these data suggest that the loss of one allele of
TSC2 is sufficient to induce morphological and physiological changes in human neurons, but only with
TSC2 biallelic mutations, a gene expression dysregulation could be detected [
86].
In PMDS, neurons derived from iPSCs displayed an impairment in excitatory synaptic transmission, depending on both a failure to generate the correct number of excitatory synapses and a decrease in the expression of glutamate receptors [
87]. Interestingly, these defects were rescued by restoring
SHANK3 expression or treating neurons with IGF-1, thus suggesting a key role of
SHANK3 in synapse formation [
87]. Moreover, PMDS iPSCs-derived neurons from fibroblasts of 2 children with ASD harboring independent de novo
SHANK3 mutations also provided a novel platform for screening active compounds able to reverse
SHANK3 haploinsufficiency by increasing SHANK3 protein levels and its recruitment to the glutamatergic synapses [
88]. The presence of synaptic abnormalities in ASD patients carrying
SHANK3 mutations was further confirmed by a recent study in which iPSCs-derived pyramidal neurons exhibited a significant decrease in dendritic spine densities, as well as in whole spine and spine head volumes [
89]. Yi and colleagues studied engineered iPSCs harboring heterozygous and homozygous
SHANK3 deletions reporting significant decreases neurite outgrowth, hyperexcitability, increased input resistance and disrupt excitatory synaptic transmission and demonstrated that these excitability deficits were at least in part due to altered surface expression of hyperpolarization-activated cyclic nucleotide–gated (HCN) channels [
90].
As already observed in iPSCs-derived NPCs, also neurons from TS patients showed defects in Ca
2+ signaling, neuronal differentiation and aberrant expression of TH that was be reversed by treatment with roscovitine, a cyclin dependent kinase inhibitor [
48]. Some authors suggested that a neuronal impairment of Ca
2+ signaling may be caused by the increased intracellular Ca
2+ influx in rodents and human iPSCs-derived neurons after membrane depolarization. Moreover, they showed that these cells exhibit the activity-dependent dendritic retraction [
91]. A recent report suggested how aberrant Ca
V1.2 splicing affects differentiation of the developing cortex during TS pathogenesis [
92].
In the first study using iPSCs-based technology for modeling Angelman syndrome (AS, OMIM #105830) and Prader-Willy Syndrome (PWS, OMIM #176270), no phenotypic differences were observed in neurons [
93]. One of the major genes implicated in ASD is
UBE3A (ubiquitin protein ligase E3A), gene involved in the AS. This disorder is typically caused by a maternal deletion within chromosome 15q11-q13, containing the gene (70 to 75% of cases). Other cases can be ascribed to paternal uniparental disomy (2% to 3%), imprinting center defect (3% to 5%), or single point mutation in the maternal
UBE3A allele (5 to 10%) [
94]. Proper gene dosage of
UBE3A is crucial to normal brain development, as evidenced by the neurodevelopmental disorders associated with this syndrome [
95]. Importantly, the authors found that
UBE3A imprinting is established during neuronal differentiation of AS iPSCs through an up-regulated expression of paternal
UBE3A antisense transcript (
UBE3A-ATS) concomitant with a repression of paternal
UBE3A [
93]. A subsequent study revealed that the silencing of paternal
UBE3A expression by
SNHG14 (also named as
UBE3A-ATS) induction is a late event during neuronal differentiation [
96]. Moreover, it has been shown that iPSCs-derived neurons from AS individuals harboring a large deletion of 15q11–q13 display impaired maturation of resting membrane potential and action potential firing, a reduction in excitatory synaptic activity and a deficit in activity-dependent synaptic plasticity [
97].