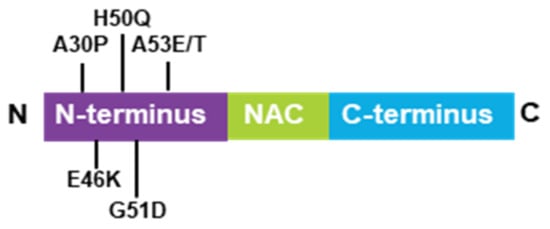
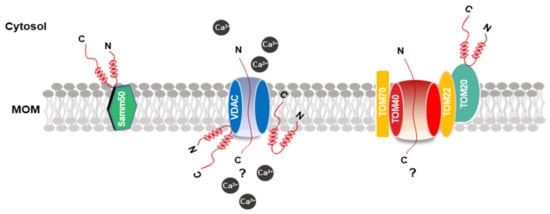
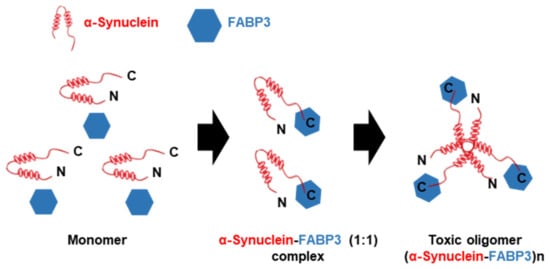
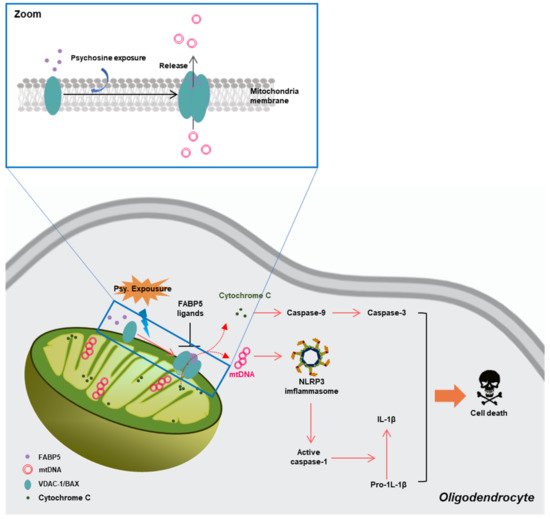
The definition of α-synuclein oligomers is rather ambiguous, owing to their heterogeneity, instability, transient existence, and variability. Oligomers are regarded as intermediates of the aggregation process but suggest higher toxicity than the other forms [
42,
43,
44] and cause a wide range of damaging effects, such as inhibition of exocytosis and dopamine release in PC12 cells [
45], complex I-dependent, Ca
2+-induced mitochondrial dysfunction in an in vitro system comprising isolated mitochondria [
46], and altering synaptic transmission in primary hippocampal and dissociated nigral neurons [
47]. Additionally, α-synuclein oligomers can permeabilize lipid bilayers [
48,
49] and alert plasma and intracellular membrane structures [
50], by rearranging the lipid microenvironment and accelerating the flux of hydrophilic molecules in the surroundings [
49,
51]. On the other hand, α-synuclein oligomers also induce pore formation and act as pathological membrane channels, while tubular amyloid or ring-shaped oligomers are integrated into the membrane [
48,
52]. Although α-synuclein oligomers seem like they connect with plasma membrane in neurons, the structural characterization of the putative pores remains unclear. Furthermore, it was reported that α-synuclein oligomers inhibit tubulin polymerization, affect the cytoskeleton of cells, and damage the integrity of the neurite network in a human dopaminergic cell line [
28,
53]. Similarly, α-synuclein overexpression also causes microtubule destabilization and neuritic degeneration in cultured cells [
54].
Misfolded and damaged polypeptides such as α-synuclein oligomers and aggregates in PD are mainly degraded by two major protein degradation systems—the autophagy-lysosomal pathway (ALP) and the ubiquitin-proteasome system (UPS) [
55]. However, in the brains of patients with PD, lysosomal activity [
56] and various lysosomal markers (LAMP1, heat-shock protein 73, and cathepsin D) were significantly decreased, especially in nigral neurons containing α-synuclein inclusions [
57]. Consistently, dementia with Lewy body (DLB) brains also exhibits ALP depletion [
58]. Additionally, α-synuclein aggregates inhibit macroautophagy by binding with essential ALP components, such as p62 and LC3 [
58]. In cultured cells, chaperone-mediated autophagy was inhibited by mutated α-synuclein expression [
59]. This inhibition results from α-synuclein binding with the specific receptor on the lysosome LAMP2, which might induce α-synuclein aggregation and injure the entire homeostasis of cells [
60]. UPS dysfunction is also present in PD brains, such as decreased proteasome enzymatic activities and structural defects of proteasomes [
61,
62], and proteasome dysfunction is considered to be of primary or secondary consequence in a wide array of chronic neurodegenerative diseases [
63,
64,
65,
66,
67]. As reported previously, both wild-type α-synuclein and mutant αSyn (A53T) expression resulted in a time-dependent and significant decrease in proteasome dysfunction in cellular models, such as SH-SY5Y cells [
68] and PC12 cells [
69]. A possible hypothesis about UPS inhibition by α-synuclein is the steric blocking of the proteasome machinery. Seventy percent of Lowy bodies, extracted from post-mortem DLB brains, are positive for 20S proteasome components [
70]; more importantly, the 20S component was found in α-synuclein aggregates extracted from pre-formed α-synuclein fibrils-treated and α-synuclein-expressing HEK293 cells [
71] and the 26S proteasome was isolated together with α-synuclein oligomers [
72]. Additionally, aggregated proteins such as amyloid-β [
70], tau [
73], and α-synuclein [
72] interact with and impair proteasome functions, and aggregated proteins such as α-synuclein and tau oligomers impair 20s proteasome function through allosteric impairment of the substrate gate in the 20S core particle and prevents the 19S regulatory particle from injecting substrates into the degradation chamber [
73,
74]. Altogether, these findings suggest that α-synuclein, especially α-synuclein oligomers, can block the UPS, thereby, damaging the clearance of other substrates and unbalancing proteostasis.