Triple negative breast cancer (TNBC) constitutes the most aggressive molecular subtype among breast tumors. Despite progress on the underlying tumor biology, clinical outcomes for TNBC unfortunately remain poor. The median overall survival for patients with metastatic TNBC is approximately eighteen months. Chemotherapy is the mainstay of treatment while there is a growing body of evidence that targeted therapies may be on the horizon with poly-ADP-ribose polymerase (PARP) and immune check-point inhibitors already established in the treatment paradigm of TNBC. A large number of novel therapeutic agents are being evaluated for their efficacy in TNBC. As novel therapeutics are now incorporated into clinical practice, it is clear that tumor heterogeneity and clonal evolution can result to de novo or acquired treatment resistance. As precision medicine and next generation sequencing is part of cancer diagnostics, tailored treatment approaches based on the expression of molecular markers are currently being implemented in clinical practice and clinical trial design.
- triple negative breast cancer
- molecular profiling
- DNA damage repair
- targeted treatment
- personalized medicine
1. Introduction
2. Molecular Profile of TNBC
2.1. Intrinsic Subtypes
2.2. DNA Damage Repair in TNBC
2.2.1. BRCA1 and BRCA2 Germline Mutations
2.2.2. BRCAness Phenotype
2.2.3. PARP Activity
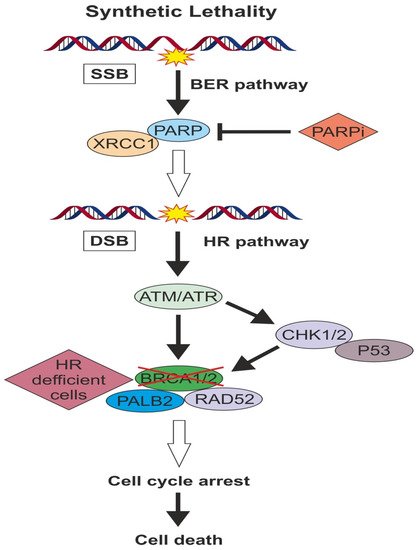
3. Targeted Pathways in TNBC
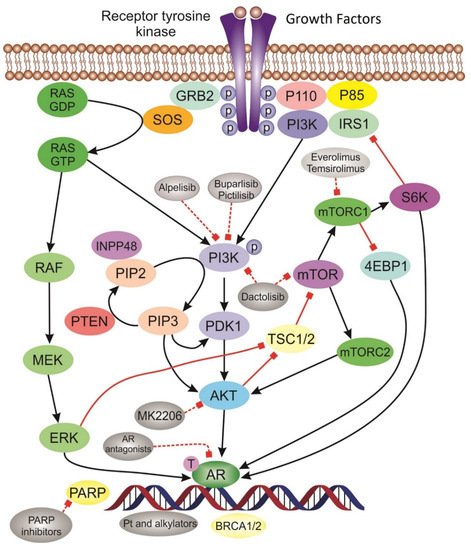
3.1. Targeting DNA Repair Pathway
3.1.1. Platinum Salts
| Trial | Design | Phase | Population | Primary Endpoint | Results | Clinicaltrials.gov Identifier |
|---|---|---|---|---|---|---|
| CALGB40603 | wPaclitaxel±Carboplatin±Bevacizumab Followed by dose dense AC vs. Standard NAC | II | Neoadjuvant Locally advanced TNBC | pCR | 62.4% vs. 22.3% | NCT00861705 |
| GeparSixto | Carboplatin+Bevacizumab+standard NAC vs. Bevacizumab+standard NAC | II | Neoadjuvant TNBC or HER2(+) | pCR | 53.2% vs. 36.9% | NCT01426880 |
| Jovanovic et al. [47] | wCisplatin+wPaclitaxel+Everolimus vs. wCisplatin+wPaclitaxel+placebo | II | Neoadjuvant Stage II/III TNBC | pCR | 36% vs. 48% | NCT00930930 |
| Zhang et al. [48] | Paclitaxel+Carboplatin vs. Paclitaxel+Epirubicin | II | Neoadjuvant Stage II/III TNBC | pCR | 38.6% vs. 14.0% | NCT01276769 |
| SHPD001 | Cisplatin (4C)+wPaclitaxel (16 weeks) | II | Neoadjuvant LABC, including TNBC | pCR | 64.7% in TNBC | NCT02199418 |
| PreECOG 0105 | Gemcitabine+Carboplatin+Iniparib | II | Neoadjuvant TNBC or BRCA1/2mt | pCR | 62.4% vs. 22.3% | NCT00813956 |
3.1.2. pCR as Surrogate Marker of Survival Benefit
3.1.3. PARP Inhibitors
| Trial | Design | Phase | Population | Primary Endpoint | Results | Clinicaltrials.gov Identifier |
|---|---|---|---|---|---|---|
| OlympiAD | Olaparib vs. PCT | Phase III | Advanced/Metastatic gBRCA, ≤2 prior lines | PFS | 7.0 vs. 4.2 months HR 0.58, p < 0.001 | NCT02000622 |
| OlympiA | Olaparib vs. placebo | Phase III | Early-stage gBRCA, adjuvant therapy | Invasive dicease free survival (IDFS) | Ongoing | NCT02032823 |
| BROCADE 3 | C + P + veliparib vs. C + P + placebo vs. Temozolamide + Veliparib | Phase II | Metastatic gBRCA, ≤0–2 prior lines lines | PFS | 14.1 vs. 12.3 months HR 0.789, p = 0.227 | NCT01506609 |
| BrighTNess | C + P + veliparib → AC vs. C + P + placebo → AC vs. Placebo + placebo + P → AC | Phase III | Stage II or III TNBC Neoadjuvant | pCR | 58% vs. 53% vs. 31% p < 0.0001 | NCT02032277 |
| I-SPY 2 | C + P + veliparib → AC vs. C + P + placebo → AC | Phase II | Stage II or III TNBC Neoadjuvant | pCR | 51% vs. 26% | NCT01042379 |
| EMBRACA | Talazoparib vs. PCT | Phase III | Advanced/Metastatic gBRCA, ≤3 prior lines | PFS | 8.6 vs. 5.6 months | NCT01945775 |
| BRAVO | Niraparib vs. PCT | Phase III | Advanced/Metastatic gBRCA, ≤2 prior lines | PFS | Completed accrual | NCT01905592 |
3.2. Antiangiogenic Agents
3.3. Immunotherapy
| Trial | Design | Phase | Population | Primary Endpoint | Results | Clinicaltrials.gov Identifier |
|---|---|---|---|---|---|---|
| Keynote-119 | Pembro vs. TPC | Phase III | Metastatic TNBC | OS | Negative | NCT02555657 |
| Keynote-355 | Pembro + chemo vs. placebo + chemo | Phase III | Metastatic TNBC 1st line | OS | Ongoing | NCT02819518 |
| Keynote-522 | Pembro or placebo + P + C × 4 followed of Pembro or placebo + AC × 4 followed of Adjuvant Pembro or placebo | Phase III | Stage II–III TNBC Neoadjuvant/Adjuvant | pCR | Interim analysis favor Pembro arm | NCT03036488 |
| Keynote-242 | Pembro vs. observation | Phase III | TNBC patients with residual disease after NACT | IDFS in ITT and PD-L1 positive | Ongoing | NCT02954874 |
| Impassion-130 | Atezo + Nab-Paclitaxel vs. placebo + Nab-Paclitaxel | Phase III | Metastatic TNBC 1st line | PFS | 7.2 vs. 5.5 months p < 0.001 | NCT02425891 |
| Impassion-030 | adj T + AC / EC vs. Atezo + T + AC / EC followed of Atezo maintenance for 1 year. | Phase III | Stage II–III TNBC Adjuvant | IDFS | Ongoing | NCT03498716 |
| Impassion-031 | Atezo + Nab-Paclitaxel + AC vs. placebo + Nab-Paclitaxel + AC followed by adjuvant Atezo | Phase III | Stage II–III TNBC adjuvant | pCR | Ongoing | NCT03197935 |
| ISPY-2 | Pembro + chemo vs. placebo + chemo | Phase III | Stage II–III TNBC Neoadjuvant | pCR | 60% vs. 20% | NCT01042379 |
3.4. PI3K/AKT/mTOR Inhibitors
3.5. Targeting AR Pathway
This entry is adapted from the peer-reviewed paper 10.3390/cancers12040916
References
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34.
- Duffy, M.J.; McGowan, P.M.; Crown, J. Targeted therapy for triple-negative breast cancer: Where are we? Int. J. Cancer 2012, 131, 2471–2477.
- Keegan, T.H.M.; Kurian, A.W.; Gali, K.; Tao, L.; Lichtensztajn, D.Y.; Hershman, D.L.; Habel, L.A.; Caan, B.J.; Gomez, S.L. Racial/ethnic and socioeconomic differences in short-term breast cancer survival among women in an integrated health system. Am. J. Public Health 2015, 105, 938–946.
- Iwata, H.; Im, S.-A.; Masuda, N.; Im, Y.-H.; Inoue, K.; Rai, Y.; Nakamura, R.; Kim, J.H.; Hoffman, J.T.; Zhang, K.; et al. PALOMA-3: Phase III Trial of Fulvestrant With or Without Palbociclib in Premenopausal and Postmenopausal Women With Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Metastatic Breast Cancer That Progressed on Prior Endocrine Therapy—Safety and Efficacy in Asian Patients. J. Glob. Oncol. 2017, 3, 289–303.
- Swain, S.; Kim, S.-B.; Cortés, J.; Ro, J.; Semiglazov, V.F.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Knott, A.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): Overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013, 14, 461–471.
- Gianni, L.; Pienkowski, T.; Im, Y.-H.; Tseng, L.-M.; Liu, M.-C.; Lluch, A.; Starosławska, E.; De La Haba-Rodriguez, J.; Im, S.-A.; Pedrini, J.L.; et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): A multicentre, open-label, phase 2 randomised trial. Lancet Oncol. 2016, 17, 791–800.
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767.
- Adélaïde, J.; Finetti, P.; Bekhouche, I.; Repellini, L.; Geneix, J.; Sircoulomb, F.; Charafe-Jauffret, E.; Cervera, N.; Desplans, J.; Parzy, D.; et al. Integrated Profiling of Basal and Luminal Breast Cancers. Cancer Res. 2007, 67, 11565–11575.
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, U.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752.
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368.
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121.
- Hilborn, E.; Gacic, J.; Fornander, T.; Nordenskjöld, B.; Stål, O.; Jansson, A. Androgen receptor expression predicts beneficial tamoxifen response in oestrogen receptor-α-negative breast cancer. Br. J. Cancer 2016, 114, 248–255.
- Wang, C.; Pan, B.; Zhu, H.; Zhou, Y.; Mao, F.; Lin, Y.; Xu, Q.; Sun, Q. Prognostic value of androgen receptor in triple negative breast cancer: A meta-analysis. Oncotarget 2016, 7, 46482–46491.
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; E Rasmussen, K.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76.
- Prat, A.; Parker, J.; Karginova, O.; Fan, C.; Livasy, C.; I Herschkowitz, J.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68.
- Atchley, D.P.; Albarracin, C.T.; Lopez, A.; Valero, V.; Amos, C.I.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Arun, B. Clinical and Pathologic Characteristics of Patients With BRCA-Positive and BRCA-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 4282–4288.
- Musolino, A.; Bella, M.A.; Bortesi, B.; Michiara, M.; Naldi, N.; Zanelli, P.; Capelletti, M.; Pezzuolo, D.; Camisa, R.; Savi, M.; et al. BRCA mutations, molecular markers, and clinical variables in early-onset breast cancer: A population-based study. Breast 2007, 16, 280–292.
- Choo, J.R.; Nielsen, T.O. Biomarkers for Basal-like Breast Cancer. Cancers 2010, 2, 1040–1065.
- Evans, D.G.; Howell, A.; Ward, D.; Lalloo, F.; Jones, J.L.; Eccles, D.M. Prevalence of BRCA1 and BRCA2 mutations in triple negative breast cancer. J. Med. Genet. 2011, 48, 520–522.
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average Risks of Breast and Ovarian Cancer Associated with BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies. Am. J. Hum. Genet. 2003, 72, 1117–1130.
- Thompson, D. Cancer Incidence in BRCA1 Mutation Carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy-a double-edged sword. J. Appl. Genet. 2019, 60, 329–334.
- Sarasin, A.; Kauffmann, A. Overexpression of DNA repair genes is associated with metastasis: A new hypothesis. Mutat. Res. Mutat. Res. 2008, 659, 49–55.
- Kastan, M.B.; Blow, J.J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323.
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113.
- Lips, E.H.; Mulder, L.; Oonk, A.; E Van Der Kolk, L.; Hogervorst, F.B.L.; Imholz, A.L.T.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177.
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ’BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120.
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.
- Pellegrino, B.; Mateo, J.; Serra, V.; Balmaña, J. Controversies in oncology: Are genomic tests quantifying homologous recombination repair deficiency (HRD) useful for treatment decision making? ESMO Open 2019, 4, e000480.
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424.
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533.
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014, 4, 1430–1447.
- Rosado, M.M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437.
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080.
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599.
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146.
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Lӧbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43.
- Kaelin, W.G. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917.
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479.
- Sikov, W.M.; Berry, D.A.; Perou, C.M.; Singh, B.; Cirrincione, C.T.; Tolaney, S.M.; Kuzma, C.S.; Pluard, T.J.; Somlo, G.; Port, E.R.; et al. Impact of the Addition of Carboplatin and/or Bevacizumab to Neoadjuvant Once-per-Week Paclitaxel Followed by Dose-Dense Doxorubicin and Cyclophosphamide on Pathologic Complete Response Rates in Stage II to III Triple-Negative Breast Cancer: CALGB 40603 (Alliance). J. Clin. Oncol. 2015, 33, 13–21.
- Von Minckwitz, G.; Loibl, S.; Untch, M.; Eidtmann, H.; Rezai, M.; Fasching, P.A.; Tesch, H.; Eggemann, H.; Schrader, I.; Kittel, K.; et al. Survival after neoadjuvant chemotherapy with or without bevacizumab or everolimus for HER2-negative primary breast cancer (GBG 44–GeparQuinto). Ann. Oncol. 2014, 25, 2363–2372.
- Jovanović, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel With or Without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045.
- Zhang, P.; Yin, Y.; Mo, H.; Zhang, B.; Wang, X.; Li, Q.; Yuan, P.; Wang, J.; Zheng, S.; Cai, R.; et al. Better pathologic complete response and relapse-free survival after carboplatin plus paclitaxel compared with epirubicin plus paclitaxel as neoadjuvant chemotherapy for locally advanced triple-negative breast cancer: A randomized phase 2 trial. Oncotarget 2016, 7, 60647–60656.
- Alba, E.; Chacon, J.I.; Lluch, A.; Antón, A.; Estevez, L.; Cirauqui, B.; Carrasco, E.; Calvo, L.; Seguí, M.A.; Ribelles, N.; et al. A randomized phase II trial of platinum salts in basal-like breast cancer patients in the neoadjuvant setting. Results from the GEICAM/2006-03, multicenter study. Breast Cancer Res. Treat. 2012, 136, 487–493.
- Von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756.
- Loibl, S.; Weber, K.; Timms, K.; Elkin, E.; Hahnen, E.; Fasching, P.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response—Final results from GeparSixto. Ann. Oncol. 2018, 29, 2341–2347.
- Von Minckwitz, G.; Hahnen, E.; Fasching, P.A.; Hauke, J.; Schneeweiss, A.; Salat, C.; Rezai, M.; Blohmer, J.U.; Zahm, D.M.; Jackisch, C.; et al. Pathological complete response (pCR) rates after carboplatin-containing neoadjuvant chemotherapy in patients with germline BRCA (gBRCA) mutation and triple-negative breast cancer (TNBC): Results from GeparSixto. J. Clin. Oncol. 2014, 32, 1005.
- Hahnen, E.; Lederer, B.; Hauke, J.; Loibl, S.; Kröber, S.; Schneeweiss, A.; Denkert, C.; Fasching, P.A.; Blohmer, J.U.; Jackisch, C.; et al. Germline Mutation Status, Pathological Complete Response, and Disease-Free Survival in Triple-Negative Breast Cancer: Secondary Analysis of the GeparSixto Randomized Clinical Trial. JAMA Oncol. 2017, 3, 1378–1385.
- Sikov, W.M.; Polley, M.-Y.; Twohy, E.; Perou, C.M.; Singh, B.; Berry, D.A.; Tolaney, S.M.; Somlo, G.; Port, E.R.; Ma, C.X.; et al. CALGB (Alliance) 40603: Long-term outcomes (LTOs) after neoadjuvant chemotherapy (NACT) +/- carboplatin (Cb) and bevacizumab (Bev) in triple-negative breast cancer (TNBC). J. Clin. Oncol. 2019, 37, 591.
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; Veer, L.J.V.T.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N. Engl. J. Med. 2016, 375, 23–34.
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; Von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509.
- Byrski, T.; Huzarski, T.; Dent, R.; Marczyk, E.; Jasiówka, M.; Gronwald, J.; Jakubowicz, J.; Cybulski, C.; Wiśniowski, R.; Godlewski, D.; et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat. 2014, 147, 401–405.
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Barni, S. The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2014, 144, 223–232.
- Gianni, L.; Pienkowski, T.; Im, Y.-H.; Roman, L.; Tseng, L.-M.; Liu, M.-C.; Lluch, A.; Staroslawska, E.; De La Haba-Rodriguez, J.; Im, S.-A.; et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): A randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 25–32.
- Baselga, J.; Bradbury, I.; Di Cosimo, S.; Aura, C.; Gomez, H.; Dinh, H.; Fauria, K.; Holmes, A.P.; deAzambuja, E.; Piccart-Gebhart, M.; et al. The association between event-free survival and pathological complete response to neoadjuvant lapatinib, trastuzumab or their combination in HER2-positive breast cancer. Survival follow-up analysis of the NeoALTTO study (BIG 1-06). In Proceedings of the SABCS, San Antonio, TX, USA, 10–14 December 2013.
- Von Minckwitz, G.; Kaufmann, M.; Kümmel, S.; Fasching, P.; Eiermann, W.; Blohmer, J.; Costa, S.D.; Sibylle, L.; Dietmar, V.; Untch, M. Integrated meta-analysis on 6402 patients with early breast cancer receiving neoadjuvant anthracycline-taxane +/- trastuzumab containing chemotherapy. Cancer Res. 2009, 69, 79.
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172.
- Tutt, A.; Ellis, P.; Kilburn, L.; Tovey, H.; Kernaghan, S.; Owen, J.; Chan, S.; Parker, P.; Perou, C.; Shaw, A.; et al. TNT: A randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer (CRUK/07/012). In Proceedings of the SABCS, San Antonio, TX, USA, 9–11 December 2014.
- Dulaney, C.; Marcrom, S.; Stanley, J.; Yang, E.S. Poly(ADP-ribose) polymerase activity and inhibition in cancer. Semin. Cell Dev. Biol. 2017, 63, 144–153.
- Michels, J.; Vitale, I.; Senovilla, L.; Enot, D.P.; Garcia, P.; Lissa, D.; Olaussen, K.A.; Brenner, C.; Soria, J.-C.; Castedo, M.; et al. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle 2013, 12, 877–883.
- Sizemore, S.T.; Mohammad, R.; Sizemore, G.M.; Yu, H.; Ostrowski, M.C.; Chakravarti, A.; Xia, F. Relationship of ionizing radiation-induced synthetic lethality of PARP inhibition in BRCA1-proficient cancer cells with p53. J. Clin. Oncol. 2016, 34, e14115.
- LaFargue, C.; Molin, G.Z.D.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28.
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.M.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533.
- Robson, M.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566.
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2018, 17, 409–419.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763.
- Poggio, F.; Bruzzone, M.; Ceppi, M.; Conte, B.; Martel, S.; Maurer, C.; Tagliamento, M.; Viglietti, G.; Del Mastro, L.; De Azambuja, E.; et al. Single-agent PARP inhibitors for the treatment of patients with BRCA-mutated HER2-negative metastatic breast cancer: A systematic review and meta-analysis. ESMO Open 2018, 3, e000361.
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416.
- Isakoff, S.J.; Puhalla, S.; Domchek, S.M.; Friedlander, M.; Kaufman, B.; Robson, M.; Telli, M.L.; Diéras, V.; Han, H.S.; Garber, J.E.; et al. A randomized Phase II study of veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in BRCA1/2 metastatic breast cancer: Design and rationale. Future Oncol. 2017, 13, 307–320.
- Miller, K.; Tong, Y.; Jones, D.R.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.-C.; Badve, S.S.; Perkins, S.M. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple negative breast cancer: Final efficacy results of Hoosier Oncology Group BRE09-146. J. Clin. Oncol. 2015, 33, 1082.
- Bi, Y.; Verginadis, I.I.; Dey, S.; Lin, L.; Guo, L.; Zheng, Y.; Koumenis, C. Radiosensitization by the PARP inhibitor olaparib in BRCA1-proficient and deficient high-grade serous ovarian carcinomas. Gynecol. Oncol. 2018, 150, 534–544.
- Shih, T.; Lindley, C. Bevacizumab: An angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 2006, 28, 1779–1802.
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E.; Robidoux, A.; Atkins, J.N.; Baez, L.; Brufsky, A.; Mehta, R.S.; Fehrenbacher, L.; et al. The effect on pCR of bevacizumab and/or antimetabolites added to standard neoadjuvant chemotherapy: NSABP protocol B-40. J. Clin. Oncol. 2011, 29, LBA1005.
- Von Minckwitz, G.; Eidtmann, H.; Rezai, M.; Fasching, P.A.; Tesch, H.; Eggemann, H.; Schrader, I.; Kittel, K.; Hanusch, C.; Kreienberg, R.; et al. Neoadjuvant Chemotherapy and Bevacizumab for HER2-Negative Breast Cancer. N. Engl. J. Med. 2012, 366, 299–309.
- Cameron, D.; Brown, J.; Dent, R.; Jackisch, C.; Mackey, J.; Pivot, X.; Steger, G.G.; Suter, T.M.; Toi, M.; Parmar, M.; et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): Primary results of a randomised, phase 3 trial. Lancet Oncol. 2013, 14, 933–942.
- Miller, K.D.; O’Neill, A.; Perez, E.A.; Seidman, A.D.; Sledge, G.W. A phase II pilot trial incorporating bevacizumab into dose-dense doxorubicin and cyclophosphamide followed by paclitaxel in patients with lymph node positive breast cancer: A trial coordinated by the Eastern Cooperative Oncology Group. Ann. Oncol. 2012, 23, 331–337.
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus Bevacizumab versus Paclitaxel Alone for Metastatic Breast Cancer. N. Engl. J. Med. 2007, 357, 2666–2676.
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370.
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients with Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467.
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.; De Laurentiis, M.; Nanda, R.; Winer, E.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411.
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443.
- Schmid, P.; Park, Y.; Muñoz-Couselo, E.; Kim, S.-B.; Sohn, J.; Im, S.-A.; Holgado, E.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Abstract PD5-01: KEYNOTE-173: Phase 1b multicohort study of pembrolizumab (Pembro) in combination with chemotherapy as neoadjuvant treatment for triple-negative breast cancer (TNBC). Poster Discuss. Abstr. 2019, 79, PD5-01.
- Schmid, P.; Cortés, J.; Bergh, J.C.S.; Pusztai, L.; Denkert, C.; Verma, S.; McArthur, H.L.; Kümmel, S.; Ding, Y.; Karantza, V.; et al. KEYNOTE-522: Phase III study of pembrolizumab (pembro) + chemotherapy (chemo) vs placebo + chemo as neoadjuvant therapy followed by pembro vs placebo as adjuvant therapy for triple-negative breast cancer (TNBC). J. Clin. Oncol. 2018, 36, TPS602.
- Nanda, R.; Liu, M.C.; Yau, C.; Asare, S.; Hylton, N.; Veer, L.V.; Perlmutter, J.; Wallace, A.M.; Chien, A.J.; Forero-Torres, A.; et al. Pembrolizumab plus standard neoadjuvant therapy for high-risk breast cancer (BC): Results from I-SPY 2. J. Clin. Oncol. 2017, 35, 506.
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. IMpassion130: Updated overall survival (OS) from a global, randomized, double-blind, placebo-controlled, Phase III study of atezolizumab (atezo) + nab-paclitaxel (nP) in previously untreated locally advanced or metastatic triple-negative breast cancer (mTNBC). J. Clin. Oncol. 2019, 37, 1003.
- Laudisi, F.; Sambucci, M.; Pioli, C. Poly (ADP-ribose) polymerase-1 (PARP-1) as immune regulator. Endocrine, Metab. Immune Disord. Drug Targets 2011, 11, 326–333.
- Aldinucci, A.; Gerlini, G.; Fossati, S.; Cipriani, G.; Ballerini, C.; Biagioli, T.; Pimpinelli, N.; Borgognoni, L.; Massacesi, L.; Moroni, F.; et al. A key role for poly(ADP-ribose) polymerase-1 activity during human dendritic cell maturation. J. Immunol. 2007, 179, 305–312.
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556.
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720.
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner Hendrickson, A.; Forero, A.; Anders, C.; Wulf, G.; et al. Abstract PD5-02: Durability of clinical benefit with niraparib + pembrolizumab in patients with advanced triple-negative breast cancer beyond BRCA: (TOPACIO/Keynote-162). Cancer Res. 2019, 79, PD5-02.
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Abstract PD5-04: An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Updated results in patients with germline BRCA-mutated (gBRCAm) metastatic breast cancer (MBC). Cancer Res. 2019, 79, PD5-04.
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619.
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Overall survival results from BOLERO-2. Ann. Oncol. 2014, 25, 2357–2362.
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940.
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399.
- Gonzalez-Angulo, A.M.; Akcakanat, A.; Liu, S.; Green, M.C.; Murray, J.L.; Chen, H.; Palla, S.L.; Koenig, K.B.; Brewster, A.M.; Valero, V.; et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancerdagger. Ann. Oncol. 2014, 25, 1122–1127.
- Martín, M.; Chan, A.; Dirix, L.; O’Shaughnessy, J.; Hegg, R.; Manikhas, A.; Shtivelband, M.; Krivorotko, P.; López, N.B.; Campone, M.; et al. A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2– advanced breast cancer (BELLE-4). Ann. Oncol. 2017, 28, 313–320.
- Kim, S.-B.; Dent, R.; Im, S.-A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372.
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel as First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2019, 38, 423–433.
- Asano, Y.; Kashiwagi, S.; Onoda, N.; Kurata, K.; Morisaki, T.; Noda, S.; Takashima, T.; Ohsawa, M.; Kitagawa, S.; Hirakawa, K. Clinical verification of sensitivity to preoperative chemotherapy in cases of androgen receptor-expressing positive breast cancer. Br. J. Cancer 2016, 114, 14–20.
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540.
- Kim, Y.; Jae, E.; Yoon, M. Influence of Androgen Receptor Expression on the Survival Outcomes in Breast Cancer: A Meta-Analysis. J. Breast Cancer 2015, 18, 134–142.
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512.
- Bonnefoi, H.; Grellety, T.; Trédan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; L’Haridon, T.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann. Oncol. 2016, 27, 812–818.
- Traina, T.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.; O’Shaughnessy, J.; Gradishar, W.J.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor–Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890.
- Traina, T.A.; Miller, K.; Yardley, D.A.; O’Shaughnessy, J.; Cortes, J.; Awada, A.; Kelly, C.M.; Trudeau, M.E.; Schmid, P.; Gianni, L.; et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). J. Clin. Oncol. 2015, 33, 1003.