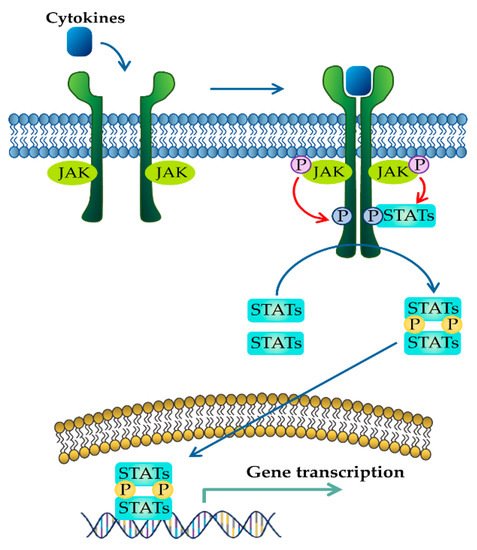
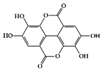
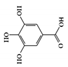
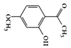
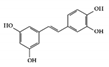
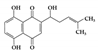
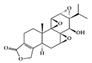
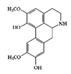
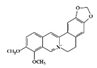
The JAK–STAT pathway is a well-conserved signaling pathway and is involved in many cellular processes, including cell division, cell death, and regulatory immune function [
27]. The JAK–STAT pathway plays a pathogenic role in many diseases, and its hyperactivation is associated with inflammatory and autoimmune diseases such as rheumatoid arthritis, IBD, systemic lupus erythematosus, and psoriasis [
28]. Although the etiology of IBD remains unknown, mucosal immune and non-immune cells in the inflamed gut of IBD patients spontaneously release pro-inflammatory cytokines such as TNF-α, interferon gamma (IFN-γ), interleukin (IL) 1 beta (IL-1β), IL-6, IL-8, and IL-12, which play a central pathologic role in IBD [
29]. These extracellular cytokines in IBD modulate inflammatory responses by activating the JAK–STAT pathway. The binding of cytokines to their cognate receptors triggers the conformational change of the receptors that alter the position of the associated JAK, resulting in phosphorylation of JAK and tyrosine residues on cytokine receptors [
30]. Phosphorylated tyrosine residues on cytokine receptors serve as binding sites for STATs, and the recruitment of STAT to the receptor induces the phosphorylation of STAT by JAK [
31]. Ultimately, phosphorylated STATs dissociate from their receptor docking sites and form homo- or heterodimers. After that, they translocate from the cytoplasm into the nucleus, where they regulate the transcription of cytokine-responsive genes () [
19]. The JAK–STAT pathway is a significant intracellular downstream signaling mediator used by various inflammatory cytokines that are increased in IBD. Thus, inhibitors targeting the JAK–STAT pathway have the advantage of suppressing multiple cytokine pathways in the treatment of IBD.
2.1. JAK Family of Proteins and JAK Inhibitors
The Janus kinase (JAK) protein family is a non-receptor protein tyrosine kinase family that includes four proteins: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). JAK3 is predominantly found in hematopoietic cells, whereas the expression of JAK1, JAK2, and TYK2 is not restricted to specific tissues [
32]. JAK is a critical intracellular signaling mediator that transduces signals from cell surface cytokine receptors to the nucleus in IBD [
33]. JAK dysregulation can result in pathological processes in IBD [
34]. In addition, various cytokines have been widely accepted as crucial inflammatory mediators leading to immunological events in IBD patients [
16]. In this regard, we focus on specific cytokines that have been reported to play a key role in IBD pathogenesis and discuss the JAKs activated by these cytokines.
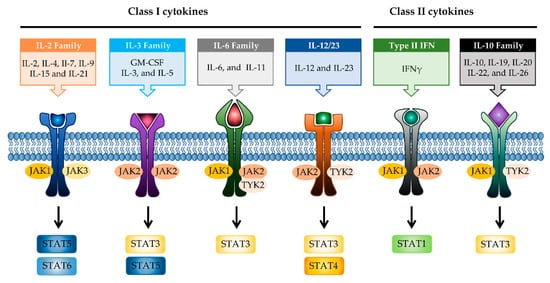
JAK differentially associates with diverse cytokine receptors activated by various cytokines and activates different types of STAT members [
19]. In other words, the JAK protein functions as a transmitter between cytokine receptors and STATs in multiple combinations, which allows the generation of specific responses to many different cytokines [
35]. Each JAK protein associates with different subunits of cytokine receptors facilitating multiple combinations with different JAK proteins, which exhibit intracellular complexity of IBD [
33,
36]. Depending on the activated signaling from specific cytokines to their cognate receptors, the pairing of JAK is determined (). Binding of IL-2 family cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21) to the type I receptor common γ-chain (γc) activates JAK1 and JAK3 [
37,
38]. Granulocyte-macrophage colony-stimulating factor (GM-CSF), which has been reported to be increased in the serum of CD patients, binds to the type I receptor β-chain and is mediated through JAK2 [
39,
40]. IL-6 has been reported to have a direct correlation with disease activity in IBD [
41]. Binding of IL-6 to type I receptor common glycoprotein 130 (gp130) primarily activates JAK1 and TYK2, followed by JAK2 and TYK2 [
42,
43]. IL-12 and IL-23 signal through the IL-12 receptor leads to the activation of JAK2 and TYK2 [
44,
45]. IL-10 and IL-22 bind to type II cytokine receptors, which activate JAK1 and TYK2 [
28]. Signaling between IFN-γ and IFN-γ receptor requires JAK1 and JAK2 [
46].
Figure 2. JAK-STAT signaling pathways in IBD. Multiple combinations with JAK proteins and STAT proteins are determined depending on the cytokines and their cognate receptors. Each cytokine family playing a key role in IBD pathogenesis is divided into two classes. JAK, janus kinase; STAT, signal transducer activator and activation of transcription; IL, interleukin; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon.
Various inflammatory cytokines are linked to the pathological effects of IBD through JAK proteins [
47]. Thus, therapy inhibiting JAK may block multiple proinflammatory cytokine signaling pathways in IBD, unlike therapy targeting cytokines or cytokine receptors. Currently, several JAK inhibitors are being evaluated for the treatment of IBD patients, and one of them, tofacitinib, has already been approved for active UC patients [
48] (). Tofacitinib is a strong selective inhibitor of JAK3 and JAK1 and has modest selectivity for JAK2 and TYK2 [
49,
50]. It can mainly block proinflammatory cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21) by inhibiting JAK1/3 and modulating other cytokines that use JAK2 and TYK2. Other JAK inhibitors are being developed in clinical trials, including peficitinib and TD-1473 as pan-JAK inhibitors [
51,
52], and filgotinib and upadacitinib with selectivity for JAK1 [
53,
54]. Although these JAK inhibitors target specific JAKs, higher doses could lead to off-target binding or immunosuppressive adverse effects [
55]. Therefore, we could alternatively consider phytochemicals, which can modulate JAK pathways in IBD, with fewer side effects than synthetic chemical drugs for the management of adverse processes related to JAK inhibition.
2.2. STAT Family of Proteins and STAT Inhibitors
The signal transducer and activator of transcription (STAT) family, which is a critical transcription factor that mediates cytokine-driven signaling, has been actively investigated in IBD pathology [
56]. The STAT protein family is composed of seven proteins: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 [
57]. Increased expression and activation of STAT1 has been reported in active IBD patients [
56,
58]. However, the function of STAT1 depends on the cell type in IBD; it is pro-inflammatory in lymphocytes and anti-inflammatory in macrophages/intestinal epithelial cells [
59,
60]. The phosphorylation of STAT1 is mediated by JAK1/JAK2 or JAK1/TYK2 and has fundamental relevance to signaling via the IFN-γ and related family of receptors [
46,
61]. STAT1 is also known to be activated by gp130 and γC family receptors [
62].
STAT2 is mostly involved in type I interferon (IFN-α and IFN-β) [
63]. Although STAT2 has been less studied in IBD pathology compared to other STAT proteins, STAT2 has been suggested to be downregulated in IBD [
56].
STAT3 has been well studied to have a fundamental role in IBD. STAT3 is phosphorylated by JAK1, JAK2, or TYK2 activated via signaling of the gp130 family of cytokines (IL-6 and IL-11) or IL-10 family members such as IL-10 and IL-22 [
64]. Several studies have reported that the expression and phosphorylation of STAT3 are increased in IBD [
65,
66]. In addition, downregulation of STAT3 has been shown to improve disease severity in a murine model of colitis [
67,
68]. The IL-6-STAT3 signaling is involved in the proliferation of lamina propria T cells and blocking of these attenuates chronic intestinal inflammation in experimental colitis [
69]. However, similar to STAT1, STAT3 is known to play a role in both pro- and anti-inflammatory effects. STAT3, which is activated by cytokines such as IL-22 and IL-10, plays a protective role in IBD. IL-22 has been reported to induce wound healing, resulting in epithelial regeneration [
70,
71,
72]. STAT3 phosphorylation by IL-10, which is produced in a wide range of innate leukocytes (macrophages, neutrophils, and dendritic cells), might play a role in preventing the disease in experimental colitis models [
73,
74]. Taken together, STAT3 promotes pro-inflammatory signals in acquired immune cells in IBD, whereas its role in innate immune cells is the suppression of colitis by enhancing mucosal protection.
STAT4 is phosphorylated by JAK2 and TYK2 in response to IL-12- or IL-23-dependent signaling [
44,
75]. STAT4 is thought to be linked to IBD based on its essential role in the function of T helper type 1 (Th1) cells, which are thought to be important for CD pathogenesis [
76]. STAT4 signaling in response to IL-12 is involved in promoting inflammatory reactions by inducing the expression of the Th1-secreted cytokine, IFN-γ [
77]. Increased expression of STAT4 in IBD patients has been shown to be involved in chronic inflammation [
78,
79]. Indeed, STAT4 knock out mice showed protective effects against experimental colitis [
80,
81]. Thus, targeting STAT4 may have therapeutic potential against IBD.
STAT5 is predominantly activated through JAK1 and JAK3 in response to the γC family of receptors by IL-2, -7, -9, -15, and -21, and is also activated by JAK2 in response to the type I receptor β-chain by the IL-3 family [
38,
82]. Several studies have shown that STAT5 plays a protective role in colitis. As a protective mechanism, STAT5 has been shown to be essential for the proliferation of intestinal epithelial stem cells, leading to the regeneration of crypt epithelium [
83]. STAT5 also plays a crucial role in IL-2 dependent forkhead box P3 (FOXP3) induction in Treg cells that can prevent intestinal inflammation in experimental colitis [
84,
85]. Thus, STAT5 may not be an appropriate therapeutic target for the treatment of IBD.
STAT6 phosphorylation arises from JAK1 and JAK3, similar to STAT5; however, it is only induced by the γC family of receptors such as IL-4R and IL-13R [
62]. STAT6 has been shown to be involved in T helper cell type 2 (Th2)-dependent IBD pathology and to have pro-inflammatory properties via the regulation of Th2 cytokines [
86,
87]. The phosphorylation of STAT6 was observably increased in the tissue of UC patients [
88,
89].
As shown above, STAT proteins could be attractive targets for the regulation of intestinal inflammation in addition to JAK for the treatment of IBD. Indeed, direct inhibitors that block STAT proteins have long been studied for treating inflammatory and autoimmune diseases, including IBD [
90]. Small molecule compounds inhibiting STAT1 signaling have been shown to improve disease in experimental colitis by selective sequestering of STAT1 from the receptor [
91]. STAT3 inhibitors reduce DNA binding of STAT3, thus blocking cell transformation [
92]. In particular, drug discovery targeting STAT3 has been extensively undertaken in various diseases, and a large amount of evidence has supported the therapeutic potential of STAT3 inhibitors [
93,
94,
95,
96]. Nevertheless, to date, there have been no direct STAT inhibitors in clinics for the treatment of IBD. It has been reported that C188-9 has preventive effects in murine IBD models [
22] (). Thus, the identification and development of phytochemicals targeting STAT proteins could have a significant role in the development of therapeutic drugs for IBD.
Table 1. Targeting the JAK–STAT pathway for IBD treatment.
| Compound |
Target |
Preclinical/Clinical Model |
Dose/Daily |
Ref. |
| JAK inhibitor |
Tofacitinib |
JAK1, JAK3 |
Approved |
10, 20 mg |
[97] |
| Filgotinib |
JAK1 |
PhaseII, III |
200 mg |
[98] |
| Upadacitinib |
JAK1 |
PhaseIII |
24 mg |
[99] |
| Peficitinib |
JAK1, JAK2, JAK3, TYK2 |
PhaseII |
25, 75, 150 mg |
[100] |
| TD-1473 |
JAK1, JAK2, JAK3 |
PhaseII, III |
20, 80, 270 mg |
[52] |
| STAT inhibitor |
C188-9 |
STAT3 |
DSS- or TNBS induced IBDmurine model |
Not designated |
[22] |