Nucleation plays a vital role in polymer crystallization, in which chain connectivity and thus the multiple length and time scales make crystal nucleation of polymer chains an interesting but complex subject. Though the topic has been intensively studied in the past decades, there are still many open questions to answer. The final properties of semicrystalline polymer materials are affected by all of the following: the starting melt, paths of nucleation, organization of lamellar crystals and evolution of the final crystalline structures. In this viewpoint, we attempt to discuss some of the remaining open questions and corresponding concepts: non-equilibrated polymers, self-induced nucleation, microscopic kinetics of different processes, metastability of polymer lamellar crystals, hierarchical order and cooperativity involved in nucleation, etc. Addressing these open questions through a combination of novel concepts, new theories and advanced approaches provides a deeper understanding of the multifaceted process of crystal nucleation of polymers.
1. What Makes Nucleation of Polymer Crystals so Difficult?
The classical path for crystallization of any substance is via nucleation and growth. The crystallization of polymers passes along the same route; an initial nucleation step (primary nucleation) is followed by a growth process. However, as shown by numerous experiments for many polymers, the connectivity of a large number of monomers in these chain-like molecules makes homogeneous nucleation to be an extremely slow process, often leading to highly unstable systems of low crystallinity prone to age and change in time. To eliminate this shortcoming of polymers, several approaches are available for enhancing the nucleation probability. For example, the use of nucleating agents [
1], self-seeding strategies [
2,
3], epitaxy [
4,
5], or the application of external forces (shearing) [
6,
7] have dramatic effects on the nucleation probability. Sometimes, blending with amorphous (incompatible) components may improve the nucleation rate [
8]. Alternatively, various thermal protocols in processing polymer systems had been established, which allowed partially or fully circumventing the nucleation step [
9,
10,
11,
12,
13], some representing unique possibilities for crystallizing polymers.
In many cases, well-established procedures used extensively for the crystallization of small molecules have been adapted and implemented for crystallizing polymer systems. However, there are also some features, which rely on the chain-like nature of polymers and thus cannot be found for systems of small molecules. Here, we would like to shed some light on already identified and potentially existing differences between small molecules and polymers with respect to their impact on nucleation mechanisms. We present and discuss a variety of mechanisms, which can initiate a polymer crystal: primary vs. secondary nucleation, self-nucleation vs. self-induced nucleation, self-seeding … We discuss how chain conformations and a change in topology in the amorphous melt may affect the mechanisms and the kinetics of nucleation of polymer crystals.
Our views are only tentative and incomplete and thus invite amendments and complementary contributions. The presented ideas are often speculative or debatable. Thus, we anticipate that our text will provoke commentaries or criticisms, which are highly welcome. A frank but respectful discussion of open questions in the field of polymer crystallization will help develop new ideas and foster new concepts.
2. Beyond Thermodynamic Concepts
Typically, most concepts of nucleation employ thermodynamic parameters to express the change in free energy between the melt and the developing nuclei. Thermodynamic principles may be justified if characteristic times for establishing (local) equilibrium are much shorter than the characteristic time of nucleation. This imposes that the process of nucleation is slower than the time required for identifying and establishing the state of the lowest free energy. At the small length-scales of the size of a nucleus, we may assume that a polymer system is at equilibrium, implying that nucleation starts from a locally equilibrated polymer melt (equilibrium conformations). Often, the final crystalline state is characterized by a stem length or a number of folds, which are also treated as a local equilibrium state having minimum free energy within this local space. However, the resulting lamellar crystals are metastable and thus may change in time with often extremely slow characteristic relaxation processes for long polymers [
14].
The formation of a nucleus for a polymer crystal requires conformational changes of the involved chains. Especially when chains interweave with others (entangled melts), equilibration processes can be very slow, and the time required for finding the state of lowest free energy for an ensemble of chains becomes increasingly long. Examples of such states have been found for melts of random copolymers [
10,
15]. For a broad spectrum of relaxation processes, polymer nucleation may become a multi-stage process. It is not obvious if conformational changes required for the formation of a (homogeneous) nucleus are the same when attaching polymers at the front of a growing crystal. Thus, differences may exist between primary nucleation and secondary nucleation.
Accordingly, we raise the following questions:
1. Do the characteristic parameters of a nucleus (e.g., shape, stem length, number of stems or chains involved …) depend on how fast the nucleus was established? For a given temperature, is it possible to define (and identify) an “equilibrated” melt state? Do we obtain different types of nuclei if polymer melts are not equilibrated? For example, is there a difference between a nucleus formed in an equilibrated melt and a nucleus formed in a non-equilibrated (e.g., sheared) melt? In this context, the observation of a “melt memory” suggests that thermal history and melt annealing time can affect characteristic parameters of a nucleus (size, shape, number of stems…). Both have been found relevant, affecting the early-stages of crystallization and hence, primary nucleation [
10,
15,
16].
2. Connectivity of the monomers represents a characteristic feature of polymer chains. Not all monomers of any long-chain (having a large number of connected monomers) are involved in the formation of a nucleus. Accordingly, if some monomers of a chain are embedded in a nucleus, what happens to the others? Is the probability of nucleation affected by the existence of a first nucleus? Is the “nucleus environment” propagating along the backbone of the polymer chains? Can the remaining molten segments form another nucleus aided by such propagation?
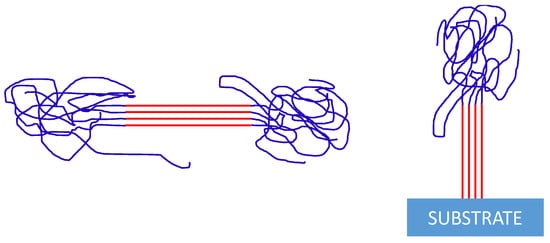
3. If monomers from several chains are integrated into a nucleus, can that lead to an enhanced probability of nucleation along the backbone of such a correlated chain ensemble? Can we establish a relation to a process, which may be called “self-induced nucleation,” on a fold surface [
17,
18,
19,
20,
21,
22]? Is it possible to draw an analogy to a bouquet of flowers, where a correlation exists between the stems (arranged in an orderly fashion) and the rather randomly arranged blooms (see )?
Figure 1. Schemes for a “bunch of flowers” of several polymer chains, which are partially involved in the formation of a nucleus (correlated stems) and partially staying amorphous (“bloom”).
Is such a coupling between crystalline and amorphous segments capable of enhancing the nucleation probability? Can the nucleation probability of (non-equilibrated) polymer chains be derived from thermodynamic arguments, or is the nucleation probability rather of a kinetic nature with rates proportional to the number of chains correlated in the “bunch of flowers”? Can we draw an analogy to sheared polymer solutions, where stretched polymer chains tend to aggregate, leading to a process similar to phase separation? (See the definition of free energy of a system of stretched chains within a mean-field approximation given by Subbotin and Semenov [
23]).
3. Nucleation from Non-Equilibrated Melts
When heating polymer crystals to a temperature above their observed melting point, often the crystallographic registry is lost, but the chain segments participating in the crystallites remain in close proximity, retaining some of the initial orientation due to a relatively slow thermal mobility. Recrystallization from such non-equilibrated melts is enhanced as the localized regions of lower entropy confer a self-nucleating melt structure. For most homopolymers, the enhanced recrystallization is limited to just 2–5 degrees above the observed melting. Crystalline melt-memory is not observed in homopolymers above their equilibrium melting temperature, and such incomplete sequence randomization in the melt disappears after increasing holding time leading to reproducible, equivalent crystallization kinetics [
24,
25,
26,
27,
28,
29]. Most experimental observations related to crystalline melt-memory in homopolymers can be explained on the grounds of thermodynamic phase behavior. Below the equilibrium melting temperature, the polymer melt is undercooled, and, besides the lack of a fast sequence diffusion and homogenization, the possibility of a small fraction of crystallites surviving in the melt cannot be excluded. In some examples, over 250 min were needed to erase self-nuclei [
29,
30].
What raises questions about the nature of such non-equilibrated melts are recent works in random ethylene 1-alkene copolymers that show memory of crystallization even at temperatures ca 30 degrees above their equilibrium melting point (~65 degrees above the observed final melting) [
10,
11,
12,
15,
16]. This unusually strong melt-memory of copolymers is in sharp contrast with the behavior of linear polyethylene fractions and was associated with the process of sequence partitioning during the copolymer’s crystallization [
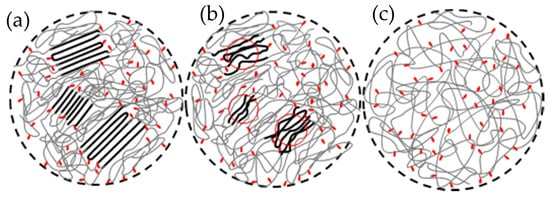
10]. As shown schematically in , due to the branches being rejected from the crystalline regions, the path of selecting and dragging ethylene sequences to build copolymer crystallites generates a complex topology of branches, knots, loops, ties and other entanglements in the intercrystalline regions, especially at high levels of transformation [
10,
31]. When the crystallites of ethylene copolymers melt, clusters from the initial crystalline ethylene sequences remain in close proximity even at very high temperatures because segmental melt diffusion to randomize all sequences is hampered by branches and the constrained intercrystalline topology (b). Melts with this type of memory are metastable systems where the clusters are effective “
pre-nuclei” that only fully dissolve into a homogeneous melt at temperatures well above the equilibrium melting point (c) [
10,
11,
12,
15,
16,
32]. Segmented thermoplastic polyurethanes are also examples of systems with selective sequence crystallization and have also shown relatively strong melt memory [
33].
Figure 2. Schematics of (
a) random copolymer semicrystalline structure with co-units (red dots) excluded from the crystalline regions; (
b) heterogeneous, non-equilibrated melt with crystalline memory: (
c) homogeneous equilibrated melt. Adapted with permission from Reference [
10]. Copyright (2013) American Chemical Society.
The experimental evidence is consistent with the kinetic nature of melt memory [
10,
11,
12,
15,
16,
24,
25,
26,
34,
35,
36,
37]. Even when cooling from the same melting temperature, the increase of crystallization temperature depends on molecular weight, the initial level of crystallinity or on how the standard crystalline state is prepared. Further studies of the effect of annealing time and molecular weight on the strong crystalline melt-memory of copolymers indicated that dissolution of such clusters, albeit thermally activated, is a very slow process. The copolymer’s melt memory persists even after > 1000 min annealing, which is unexpected on the basis of prior self-diffusion and melts relaxation times for the same copolymers [
15]. The very long characteristic times associated with the dissolution of melt memory observed for HPBDs contrast with relaxation times extracted from various avenues for the same systems [
15]. For example, prior work on melt diffusivity for HPBDs at temperatures between 140 and 180 °C with a molecular weight ranging from 10.000 to 500.000 gmol
−1 estimated single-chain relaxation times between 1 ms and 10 s [
38,
39,
40,
41]. These values are 3–7 orders of magnitude smaller than the characteristic time for dissolution of memory. Hence, dissolution of melt memory appears to entail more than just reptation of polymer chains because the process of diffusing and randomizing ethylene segments from the initial crystallites is a lot costlier than classical translational chain diffusion. Such a discrepancy points to a dissolution process of memory that is not correlated with the single-chain dynamics.
What is the topology of the chain segments that emanate from the core crystalline lamellae that make the dissolution of memory (pre-nuclei) such a slow process? This feature must be explained beyond thermodynamic concepts, as mentioned earlier. Could such non-equilibrated melts be considered as metastable states that may have originated during crystallization as posited? [
42] While testing the latter may be feasible with reliable isothermal experimental data, the small mass fraction involved in efficient self-nuclei in melts with memory may hamper direct experimental detection.
This entry is adapted from the peer-reviewed paper 10.3390/cryst11030304