2.1. Hypoxia-Inducible Factors
HIFs are the predominant mediators of a cell’s metabolic and physiological response to hypoxia and have increasingly been found to influence EMT, metastasis, and chemoresistance. HIF signaling can be activated by the PI3K, AKT, MAPK, and NF-ĸB pathways which can be activated by cytokines such as TNF-α, chemokines, G protein-coupled receptors, Toll-like receptors, and other factors [
5]. In triple-negative breast cancer (TNBC), HIF-1 accumulation occurs as a result of glutamate secretion which inhibits the xCT glutamate-cystine antiporter leading to intracellular cysteine depletion [
107]. The absence of cysteine under hypoxia subsequently inactivates prolyl hydroxylase EglN1 which usually facilitates HIF-1α degradation in normoxia [
107]. Other pathways such as PI3K/AKT signaling, in conjunction with a HIF-1-induced transcriptional response, induced cisplatin chemoresistance and EMT marker expression in hepatocellular carcinoma (HCC) cells [
108]. Numerous associations have been highlighted between HIF levels and metastasis, disease recurrence, and poor prognosis in ovarian, breast, thyroid, and lung cancers [
109,
110,
111,
112]. HIF-1α expression is associated with reduced disease-free survival and a poorer response to hormone-based therapy in breast cancer [
113].
In hypoxic conditions, HIFs are stabilized because of the inhibition of oxygen-dependent dioxygenases which modify HIFs under normoxia, enabling its degradation. The stabilization of HIF allows for binding at consensus sequences on DNA and the recruitment of p300, RNA polymerase II, and cofactors so that target genes are transcribed. The downstream effects of increased HIF signaling include regulation of the expression of genes involved in glucose metabolism, erythropoiesis (
EPO), vascularization (
VEGF,
SDF1,
KITL), tissue remodeling (
LOX), and wound healing (
TGFA) [
114]. Chromatin immunoprecipitation assays reveal over 1,000 genes regulated by HIF-1 [
115], but only a subset of the extensive transactivation, usually 100 genes, is typically observed in a given cell [
116].
While there is substantial sequence and structural homology between HIF-1 and HIF-2, they carry out different functions. HIF-1α is the major factor that contributes to target gene transactivation, tumorigenesis, and metastasis in the TNBC cell line, MDA-MB-435 [
117]. Both HIF-1α and HIF-2α activate VEGF and form complexes with the c-Myc oncoprotein to regulate transcription [
118]. However, HIF-1α inhibits c-Myc activity, whereas HIF-2α potentiates it [
119]. In addition, HIF-1α uniquely activates glycolytic enzymes phosphoglycerate kinase (PGK) and aldolase A (ALDA), glucose transporter GLUT1 (or SLC2A), stem cell marker OCT4, the pH regulator, carbonic anyhydrase IX (CA9), and EMT-associated transcription factors Zeb, Snail, and TWIST [
120,
121,
122]. Specifically, CA9 is a prognostic factor, hypoxic indicator, and promising therapeutic target, as inhibiting CA9 can enhance anti-PD-1 and anti-CTLA-4 blockade in melanoma and breast cancer models [
123]. In addition, additional targets of HIF-1α include the Wnt and Notch pathways which contribute to EMT, and LOX which activates Snail, represses E-cad, and contributes to chemoresistance in TNBC [
121,
124,
125,
126]. Interestingly, HIF-1α but not HIF-2α appears to regulate extracellular acidification in hypoxic tumors while both isoforms contribute to radioresistance in NSCLC [
127]. HIF-2 has been identified as the principal oncogenic HIF isoform in clear cell renal cell carcinoma (ccRCC), contributing to a “pseudo-hypoxic” cell state, and has been implicated in pathogenesis, EMT, and angiogenesis in NSCLC [
128]. HIF-2 controls cell differentiation and adaptive responses to hypoxia [
129,
130], along with erythropoiesis [
131,
132]. The functions of HIF-3α are more elusive due to multiple splicing variants that make functional characterization of the isoform challenging in research. HIF-1α expression can be induced by nitric oxide and ROS [
133], but there are also hypoxia-independent mechanisms of regulating HIF by the PI3K/AKT/mTOR pathway, cytokines, epigenetic changes, and lipopolysaccharides, which are not discussed in this review [
134].
Recent research has revealed the intersectionality between HIF-1, angiogenic signals, and metastasis. HIF-1 is known to promote vascularization of tumors through the upregulation of VEGF, which in one mechanism, binds to VEGFR2 receptors on bone marrow cells (BMCs), mobilizing the cells to promote angiogenesis [
135]. The anthracycline doxorubicin prevents angiogenesis and tumor growth by reducing HIF-1 signaling and the circulation of these BMCs [
135]. Similarly in melanoma and lung cancer, colonizing tumor cells produce VEGF-A, LOX, TNF-α, and inflammatory serum amyloid A3 in a HIF-1-dependent manner which leads to the recruitment of bone marrow-derived cells, metastatic niche formation, and remodeling of the extracellular matrix to facilitate invasion [
136]. HIF-1α also regulates extracellular matrix metalloproteinase inducer which is induced by hypoxia and promotes metastasis and EMT in esophageal cancer cells [
137]. HIF-1 also induces the expression of L1 cell adhesion molecule which allows breast cancer cells to adhere to blood vessel endothelial cells and metastasize to the lung [
138]. A study using a mouse model of melanoma found that inactivation of HIF-1α or HIF-2α had no change in tumorigenesis but significantly reduced metastasis, suggesting a specific function of HIF in metastasis [
139]. Interestingly, in MDA-MB-231, a human TNBC cell line, VEGF-D is inversely correlated with hypoxia and metastasis to lymph nodes, while platelet-derived growth factor B (PDGF-B) was found to be directly activated by HIF-1 and to facilitate lymphatic metastasis [
140].
Glycolysis-related HIF-1-transactivated genes might also be a driver in metabolic reprogramming that occurs during metastasis; for instance, HIF-1 activates monocarboxylate transporter 4 (MCT4) expression which promotes the transport of lactate into the ECM, acidifying the TME and favoring pre-metastatic remodeling of ECM [
141]. In addition, the metastatic site likely selects for particular metabolic and enzymatic profiles that enable tumor cells to survive. Pyruvate dehydrogenase kinase (PDK1) and glycolytic metabolism facilitated breast metastases to the liver more so than to the bone or lung [
142].
In addition to the altered milieu of intracellular and extracellular metabolites, hypoxic tumors develop mechanisms that resist the antitumor immune response. The HIF-1-mediated production of TGF-β, VEGF, and CCL28 contributes to immunosuppression in the TME via the recruitment of regulatory T cells (T
regs), macrophages, and myeloid-derived suppressor cells [
143]. T
regs are partially responsible for tumor immune tolerance, angiogenesis, and metastasis. Additionally, adenosine production and secretion, associated with an immunosuppressive environment, is increased under hypoxia as a result of the HIF-1α mediated upregulation of CD73 and CD39 [
144]. HIF-1 upregulates PD-L1 expression in tumors leading to the overstimulation and exhaustion of T cells, blocking their cytotoxic functions [
145].
Hypoxia enriches a chemo-resistant CSC population in TNBCs treated with cytotoxic paclitaxel or gemcitabine both in vitro and in vivo through HIF-1α expression [
146]. The increase in proportion of breast CSCs was coincided with increased IL-6, IL-8, MDR1, and ROS, and this expression was inhibited with the coadministration of HIF inhibitors digoxin or acriflavine [
146]. Both HIF-1α and HIF-2α are known to activate Notch signaling and “stemness” and EMT transcription factors [
62,
147] which can consequently interact with the Wnt pathway and regulate the stem cell phenotype. Leukemic cells that have adapted to hypoxia exhibit stem cell-like properties along with increased HIF-1α, β-catenin, and glyoxalase-1 activity, an enzyme that detoxifies the harmful glycolytic by-product, methylglyoxal [
148]. In this way, hypoxia-induced metabolic, transcriptional, and molecular alterations converge enabling subpopulations of treatment-resistant, stem cell-like cancer cells to survive and contribute to disease relapse.
HIFs drive processes that affect metabolism and cellular behavior, but conversely, EMT and environmental factors such as drug administration can affect cancer metabolism and transcriptional responses. Despite the pervasive influence of HIF pathways on the hypoxic response, some HIF-independent mechanisms for metastasis, EMT, and resistance to therapy may exist in different cancer types and stages.
2.2. Oxoglutarate-Dependent Dioxygenases
2OGDDs are a diverse superfamily of enzymes (e.g., prolyl hydroxylases, JmjC (Jumonji C) domain histone lysine demethylases (KDMs), ten-eleven translocation (TET) DNA hydroxylases, and RNA demethylases such as FTO and ALKBH1-3, 5) that facilitate numerous biological processes, including the HIF-mediated response to hypoxia, ECM formation, DNA and histone modifications, and normal and cancer cell metabolism. 2OGDDs hydroxylate their substrate with the assistance of oxygen, 2-oxoglutarate, and Fe
2+ and produce succinate in the reaction [
15]. In the canonical response to hypoxia, a specific class of 2-OGDDs, prolyl hydroxylase domain proteins, hydroxylate prolyl residues of HIF (Pro402 and Pro564 in HIF-1α; Pro405 and Pro531 in HIF-2α; Pro492 in HIF-3α) which enable the von Hippel-Lindau protein to ubiquitinate HIF-α and tag it for degradation by proteasomes [
13]. While dysregulated 2OGDDs have been implicated in many cancers, cancer-related metabolic alterations can also influence 2OGDDs. PHDs (or EglNs) are inhibited by oncometabolites succinate and fumarate, which accumulate under hypoxia, and 2OGDDs can also be targets of HIF signaling.
Various 2OGDDs have been linked to the development of the CSC phenotype and chemoresistance. PHD2 (EglN1) is inhibited by TGF-β a major factor for EMT and metastasis, resulting in the stabilization of HIF-1 and the enhancement of EMT pathways [
149]. In addition, PHD1 (EglN2) inhibition and silencing sensitizes colorectal cancer cells to 5-FU chemotherapy [
150]. KDM5A and KDM5B are associated with transcriptomic heterogeneity and therapy resistance in luminal breast cancer and melanoma [
151,
152]. Loss of KDM6A, a known tumor suppressor, contributes to histone hypermethylation, a phenomenon common in hypoxic tissues, and was found to prevent cellular differentiation under hypoxia, independently from HIF [
153]. Likewise, KDM6A and KDM5C are often mutated in ccRCC (3% and 8% of tumors, respectively) [
154,
155]. KDM6A is also mutated in numerous solid tumors such as bladder cancer, prostate cancer, and breast cancer [
156], and depletion of KDM6A led to increased expression of EMT transcription factors, Snail, ZEB1, and ZEB2 [
157]. ALKBH5, an m6A RNA demethylase, is responsible for a HIF-1-mediated increase in NANOG expression and the induction of a CSC phenotype in breast cancer [
158]. Overall, targeting oncogenic 2OGDDs has potential to not only inhibit tumor growth and CSC features but also enhance existing anticancer therapies.
2.3. The Unfolded Protein Response Pathway
Oxygen is a necessary electron acceptor to facilitate disulfide bond formation in protein folding, but under severe hypoxia, impaired protein folding, along with HIF-1-related metabolic switches, mitochondrial stress, or ROS activity can activate the unfolded protein response (UPR) pathway [
159]. Although the UPR signaling cascade typically promotes cell survival and adaptation to hypoxia by regulating protein production, degradation, and cell metabolism, it can also induce cell death [
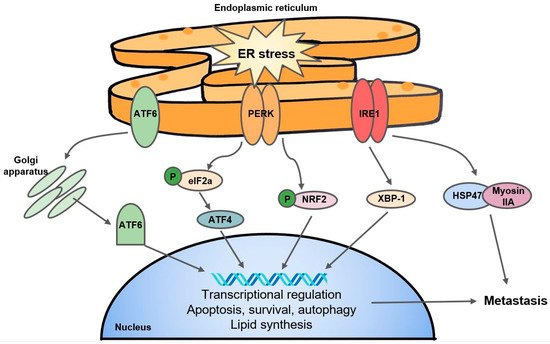
160]. The UPR can be characterized by three major endoplasmic reticulum (ER) stress sensors: the PRKR-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1), and the ATF6 pathway () [
161]. Together, to mediate ER stress, UPR signals transiently suppress mRNA translation and protein biosynthesis and activate protein degradation and cellular apoptosis [
161].
Figure 3. A simple schematic of the UPR. The three major arms of the UPR consist of ATF6, PERK, and IRE1. ATF6: activating transcription factor; PERK: PRKR-like endoplasmic reticulum kinase; IRE1: inositol-requiring enzyme 1; eIF2a: eukaryotic initiation factor 2a; NRF2: Nuclear factor erythroid 2-related factor 2; XBP-1: X-box binding protein 1; HSP47: heat shock protein 47.
Mechanistically, PERK interacts with and phosphorylates eukaryotic initiation factor 2a (eIF2a) which regulates translation of downstream effectors such as activating transcription factor 4 (ATF4). The PERK-ATF4 branch is known to trigger CREB3L1 activity which contributes to metastasis and EMT in breast cancer [
162]. The PERK-eIF2a arm of the UPR has been linked to hypoxic cancer cell survival and tolerance to radiotherapy in a colorectal cancer cell line through a mechanism of glutathione synthesis and mitigation of the effects of ROS [
163]. Separately, Yoneda et al. demonstrate that IRE1, another UPR transducer, facilitates the interaction of HSP47, a chemical chaperone, with non-muscle myosin IIA, contributing to aggressive metastasis in breast cancer [
164]. The expression of XBP-1, another substrate of IRE1, was found to drive TNBC growth and invasiveness, and correlated with hypoxia-mediated HIF-1α gene signatures [
165]. Interestingly, the IRE1-XBP-1 axis is also responsible for the downregulation of c-Myc and activation of NK cells which protected against melanoma [
166]. In addition, the fact that UPR inhibitors such as 4-PBA and TUD-CA can stall tumor growth and metastasis [
167] suggest that the UPR pathway may present as a potential anticancer target. Interestingly, researchers have found that KDM1A inhibitors are able to induce differentiation in glioma stem cells by activating the UPR [
168], and ATF4 stability is mediated by PHD3 [
169], speaking to the complex and understudied overlap of 2OGDDs with the UPR.
2.4. Other Emerging Pathways, from Exosomes to Noncoding RNAs
Exosomes or microvesicles are nanosized vesicles secreted extracellularly that aid in cell–cell communication and sculpting the TME. Able to carry proteins, lipids, microRNAs, or mRNAs, exosomes have been associated with cancer progression, angiogenesis, and EMT. Hypoxia induces the increased release of exosomes in glioblastoma cells along with ovarian, breast, and prostate cancer cells [
170,
171,
172,
173]. Hypoxia also influences the composition of molecules within exosomes, such as increasing amounts of triglycerides, metalloproteinases, IL-8, LOX, and heat shock proteins, and inducing the exosomal secretion of miRNAs that induce angiogenesis and stemness [
170,
174,
175,
176]. Ovarian cancer cells upregulate the exosomal efflux of cisplatin when treated with the drug, and inhibiting exosome release using Amiloride impaired tumor cell proliferation [
173]. Exosomes derived from CSCs promote the survival of immunosuppressive neutrophils, ultimately accelerating colon cancer growth [
177]. The secretion of exosomes may in part be mediated by HIF-1 [
178,
179]. Overall, these data suggest that hypoxia-induced exosomes may contribute to tumorigenesis and chemoresistance.
Mitochondrial dynamics such as mitochondrial fission and motility influence cell survival, morphology, and ROS homeostasis which may affect EMT and metastasis. For instance, the distribution and motility of mitochondria which is dependent on MIRO1 and MIRO2, Rho-GTPases that regulate mitochondrial movement through anchorage to kinesin or dynein, has been found to influence cancer metastasis [
180]. In addition, Dynamin related protein 1 (Drp1) facilitates mitochondrial fission and is upregulated by hypoxia in MDA-MB-231 TNBC cells; silencing of Drp1 reduced mitochondrial fission, ROS production, apoptosis, and migration in TNBC cells [
181]. While there is evidence linking Drp1 with stemness in ER-positive breast cancer cells [
182], there is a need for additional research to definitely link mitochondrial fission and other dynamics with the CSC phenotype. Previously, our research indicated that PHD1 (EglN2) promotes the binding of peroxisome proliferator-activated receptor-γ coactivator (PGC1α) with NRF1 under hypoxia and subsequently maintains microchondrial biogenesis in breast cancer through inducing transcription of ferridoxin reductase (FDXR) [
183]. Recently, protein-tyrosine phosphatase mitochondrial 1 (PTPMT1), an enzyme essential for cardiolipin biosynthesis and mitochondrial membrane integrity, was identified in a genome-wide CRISPR-Cas9 knockout library screening as a crucial survival factor for HCC cells under hypoxia [
184].
PTPMT1 regulates cardiolipin synthesis and facilitates the assembly of the ETC complexes which alleviates ROS accumulation during hypoxia. Depletion or pharmacological inhibition of PTPMT1 decreases tumor growth, disrupts the mitochondrial membrane and ETC formation, and reduced metastasis in different cancers [
184]. In addition, hypoxia-induced mitochondrial stress is a hallmark of intra-tumoral T cells with persistent antigen stimulation. These exhausted T cells have repressed PGC1α and are less able to mitigate the effects of ROS, suggesting that hypoxia-induced ROS might be connected to T cell exhaustion and dysfunction [
185]. By targeting and reversing hypoxia, terminal T cell exhaustion can be prevented, increasing the efficacy of checkpoint blockade immunotherapy [
185].
The effects of microRNAs (miRNAs) on metastasis, the CSC phenotype, and resistance to antitumor therapies are an understudied field. MiRNAs, while too short to encode proteins themselves, can inhibit the translation of mRNA or facilitate the degradation of target mRNA. With the potential to regulate the expression of a variety of proteins, miRNAs can be tumor suppressive or oncogenic. Hypoxia-mediated miRNA, miR-210, was upregulated in the CSC subpopulation of MCF-7 breast cancer cells, and suppressed E-cadherin and upregulated Snail expression in the breast CSC population [
186]. On the other hand, hypoxia-inducible miR-155 repressed homology-directed repair factors such as RAD51 in breast cancer cells and enhanced sensitivity to irradiation [
187]. A review paper provides a list of miRNAs that contribute to resistance to chemotherapy agents in many cancers including those of the breast, ovary, stomach, colon, and lung [
188].
Several studies have pointed out the role of long noncoding RNAs (lncRNAs), noncoding strands of RNA longer than 200 nucleotides, in regulating cancer development. A substantial proportion of single-nucleotide polymorphisms linked to risk in cancers are encoded on lncRNAs, and like miRNAs, lncRNAS can also regulate mRNA translation and degradation [
189]. LncRNAs have been found to promote metastasis in OSCC by operating through pathways known to induce EMT such as the AKT, Wnt, and NF-ĸB pathways [
190,
191,
192]. Recently, it was discovered that RAB11B-AS1, a lncRNA transcriptionally induced by HIF-2, promotes angiogenesis and metastasis in TNBC tumors grown in mice through the upregulation of VEGFA and ANGPTL4 [
193].
The relationship between circular RNA, defined as single-stranded noncoding RNA that forms a continuous loop via a covalent bond between 3′ and 5′ ends, and cancer is a less studied field. One study reports that circHIPK3, mediated by HIF-2α, contributes to metastasis and invasion in hypoxia-adapted gastric cancer cells through the interaction with miR-653-5p and miR-338-3p and subsequent activation of the AKT pathway [
194].
Histone deacetylases (HDACs) are a group of enzymes which modify chromatin structures by removing acetyl groups from lysine residues in histones and transcription factors and can therefore directly contribute to epigenetic and transcriptional alterations during cancer adaptation and survival under hypoxia. HDACs are frequently overexpressed across many cancer types and have been implicated in angiogenesis and cancer proliferation. HDAC1-3 repress miRNA-449 in HCC cells, allowing tumorigenic c-MET to promote growth signals [
195]. HDAC6 enables α-tubulin deacetylation in hypoxic conditions, allowing EMT factor, SMAD3, to translocate to the nucleus; HDAC6 inhibitors have been shown to inhibit metastasis in TNBC and angiogenesis in gastric cancer cells by reducing HIF-1α and VEGF levels [
196,
197]. A recent study reveals a novel role for HDAC6 in glycolysis, and inhibition of HDAC6 not only decreases growth and invasion in TNBC but directly increases acetylation of glycolytic enzymes such as GAPDH, aldolase, and enolase [
198].
Another outcome of hypoxia is autophagy, in which cellular stress induces the lysosomal degradation and recycling of proteins and damaged organelles into nutrients to maintain cell functions and promote survival. Together, HIF-1 signaling, hypoxia-mediated metabolic reprogramming, the UPR, and mTOR signaling converge on autophagy and ultimately contribute to tumor proliferation and metastasis [
199]. Autophagy can be dependent on or independent of HIF-1 signaling, but it has been shown that HIF-1-mediated upregulation of autophagy genes beclin1 (BECN1) and ATG5 enabled lung tumor immune evasion [
200]. In addition, BECN1 was responsible for an impaired natural killer cell-mediated antitumor immune response in breast cancer [
201].
From miRNAs, lncRNAs, exosomes, and the UPR to the complex transcriptional responses induced by HIFs, hypoxia ignites an avalanche of responses that reconfigure cell metabolism, local tumor immunity and vasculature, and sensitivity to additional stressors such as chemo- or radiotherapy. The co-regulation, convergence, and interdependence of multiple pathways within a hypoxic tumor have great implications in the development and enhancement of anticancer treatments.