Poly (ADP-ribose) polymerases (PARP) 1-3 are well-known multi-domain enzymes, catalysing the covalent modification of proteins, DNA, and themselves. They attach mono- or poly-ADP-ribose to targets using NAD+ as a substrate. Poly-ADP-ribosylation (PARylation) is central to the important functions of PARP enzymes in the DNA damage response and nucleosome remodelling. Activation of PARP happens through DNA binding via zinc fingers and/or the WGR domain. Modulation of their activity using PARP inhibitors occupying the NAD+ binding site has proven successful in cancer therapies. For decades, studies set out to elucidate their full-length molecular structure and activation mechanism.
1. Introduction
Poly ADP-ribose polymerase (PARP) enzymes play a key role in a number of cellular processes, such as DNA repair, genome maintenance, and cell death [
1,
2,
3,
4,
5,
6,
7]. The best characterised member of the PARP family is PARP1, which was first identified for its role in the recognition and repair of single-strand DNA breaks (SSB) [
8,
9]. Since then, PARP1 has also been shown to have a role in a number of DNA damage response (DDR) pathways, including base excision repair (BER), homologous recombination (HR), non-homologous end joining (NHEJ), and DNA mismatch repair. Consequently, PARPs have been an attractive target for anti-cancer therapies resulting in the successful development of several PARP inhibitors (PARPi) for ovarian, breast, lung, and pancreatic cancers [
10]. This novel class of therapies competes with the native substrate nicotinamide adenine dinucleotide (NAD
+) for the PARP catalytic site [
11,
12,
13,
14,
15].
It has been more than half a century since the discovery of PARP1 and the process of poly-ADP-ribosylation (PARylation) that PARP1, and other members of the PARP family, catalyse [
1,
2,
3]. During PARP catalysis ADP-ribose residues are transferred from NAD
+ onto the target substrate building a poly ADP-ribose (PAR) chain. The building of PAR chains, along with their removal, occurs in all three major divisions of life (eukaryotes, prokaryotes, archaea) [
16,
17,
18]. Here, we discuss the human enzymes in the context of DNA damage repair. The identification of PAR and its structure, and the realisation that PARP1 produces PAR, were key breakthroughs during early PARP research [
19,
20,
21,
22]. Studies that followed detailed the purification of PARP1 [
23], demonstrated the activation of PARP in response to genotoxic agents [
8,
24], linked PARP1 to the repair of DNA damage [
9], and showed the association of PAR with nucleosome remodelling and enzymes, including PARP1 itself [
21,
25]. Furthermore, the discovery of PARP2—and other members of the PARP family—was enabled by the generation and characterisation of the
PARP1 knockout mouse [
26,
27,
28].
The PARP family consists of 17 proteins in humans. PARP1, PARP2, PARP5A, and PARP5B are capable of synthesising PAR chains [
1,
2,
3]. Most other members in the PARP family catalyse the addition of single ADP-ribose units and are therefore classified as mono ADP-ribosyltransferases (MARs). PARP1, PARP2, and PARP3 are DNA-dependent enzymes [
4]. PARP1, the largest of the three, is approximately 116 kDa [
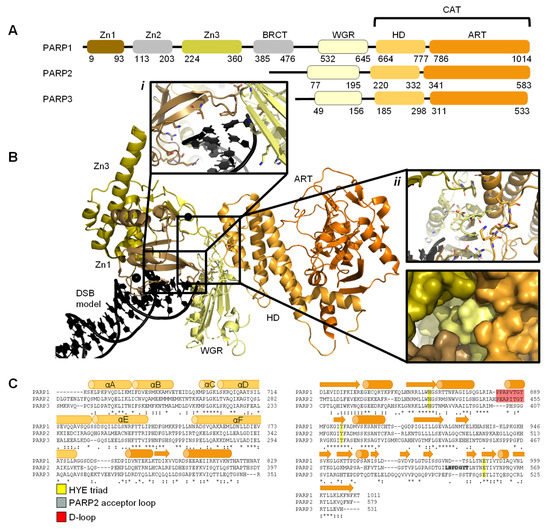
29] and comprises of six independently folded domains: the N-terminus consists of three zinc (Zn) finger domains Zn1, Zn2, and Zn3; this is followed by the auto-modification domain, which contains the BRCA1 C-terminus (BRCT) fold and mediates protein–protein interactions; adjacent to this is the tryptophan, glycine, arginine (WGR) motif; whilst at the C-terminus sits the catalytic (CAT) domain () [
30,
31,
32,
33]. Four of these domains (Zn1, Zn2, Zn3, and WGR) have been shown to bind DNA (B) [
34]. The CAT domain is the most conserved across the PARP family and comprises of the helical subdomain (HD) and the ADP-ribosyl transferase (ART) subdomain (C) [
30,
31]. Interestingly, PARP2 and PARP3 only share the C-terminal regions (WGR and CAT domains), yet they are able to regulate the mechanism of DNA-induced activation via local destabilisation of the HD. It has been proposed that human PARP2 contains an additional N-terminal DNA and/or RNA-binding domain [
35,
36,
37]: the SAP motif named after the proteins in which it was found, SAF/Acinus/PIAS [
38]. This motif occurs one to four times at the N-terminus of plant PARP2 [
39,
40] and is a putative DNA-binding four-helix bundle [
41]. However, Riccio et al. (2015) [
42] report that although the N-terminus of PARP2 is important for PARP2 activation on SSBs, the N-terminal region of human PARP-2 is intrinsically disordered [
42]. Therefore, here the SAP motif is not included in the domain overview of PARP2 (A).
Figure 1. Overview of PARP1, PARP2, and PARP3. (
A)
. Schematic overview of the domains of PARP1, PARP2, and PARP3, with amino acid numbers indicating domain boundaries corresponding to Uniprot entries P09874, Q9UGN5, and Q9Y6F1; (
B)
. Crystal structure of PARP1 bound to a double-strand DNA molecule representing one end of a double-strand break (DSB) (Protein Data Bank (PDB) entry 4DQY [
34]). Domain colours correspond to A. Inset
i: interactions of Zn1 and WGR with a DSB model. Inset
ii: interdomain interactions between Zn1, Zn3, WGR, and HD shown as cartoons (top) and surface representations (bottom). All figures in this review containing protein structures were generated in PyMOL [
43]; (
C)
. Structure-based sequence alignment of CAT domains of PARP1, PARP2, and PARP3 was generated in PyMOL [
43]. Sequence alignment was generated in Clustal Omega [
44] provided by EMBL-EBI [
45]. Secondary structure is represented by cylinders as α-helices and arrows as β-sheets, respectively. Sequence conservation is shown, where an asterisk represents fully conserved, a colon represents strongly similar properties and a period represents weakly similar properties. The conserved histidine, tyrosine, glutamic acid (HYE) triad is shown in yellow, the PARP2 acceptor loop in grey, and the D-loop in red.
Early structures of individual PARP domains were first published in the late 1990s for PARP1 [
46,
47,
48], and early 2000s for PARP2 [
49]. The first crystal structure of the human PARP1 CAT domain bound to a small molecule inhibitor was published by Kinoshita and co-workers in 2004 [
50], and subsequently, for PARP2 by Karlberg et al. (2010) [
51] and for PARP3 by Lehtiö and colleagues [
52]. Thereafter, many more structures have been published of PARP1-3, often of individual domains and short truncated forms of the proteins. To date there have been no reported structures of full-length PARP1. However, the full-length structure of PARP2 and histone PARylation factor 1 (HPF1) in complex with a nucleosome was recently determined by cryo-electron microscopy (cryo-EM) [
53].
2. DNA Damage Recognition by PARP Enzymes
Each cell within an organism faces a high frequency of DNA damage on a daily basis [
73,
74], due to many endogenous and exogenous causes [
75]. Members of the PARP family are key initiators of the DDR pathway amongst other functions [
4,
6,
76]. Repairing damaged DNA is essential for cells to enable successful transcription, maintain genomic stability, achieve cell replication, and survive [
75]. The majority of DNA damage within cells is inflicted on just one DNA strand and is known as a single strand break (SSB). These SSBs are often easier to repair given the necessary information is still available on the complementary strand. Different types of SSB damage can occur depending on the process which resulted in the break. For example, a DNA strand can be nicked (both bases are intact but the DNA backbone is broken), the pyrimidine/purine group can be missing resulting in an abasic (AP) site, or a nucleotide can be missing (gap) [
73]. Double-strand breaks (DSBs) are more problematic for cells and therefore cells have dedicated pathways to rectify this type of damage. Depending on the cell cycle stage, and the presence of a template, the HR or the NHEJ pathways are initiated [
10,
77].
PARPs play an essential role as DNA damage sensors, of both SSBs and DSBs [
5], and promote DNA repair through recruitment of DNA repair factors. Examples of such PARP1 interactors include: the X-ray repair cross complementing group 1 (XRCC1) [
78] acting as a scaffold for DNA SSB repair components; the tyrosyl-DNA phosphodiesterase 1 (TDP1) [
79], which removes stalled topoisomerase 1 (TOP1)-DNA complexes; DNA protein kinase catalytic subunit (DNAPKcs) [
80] in the NHEJ DSB repair pathway and variable, diversity, and joining (V(D)J) recombination [
81]; the kinase ataxia telangiectasia mutated (ATM) [
82,
83] in DNA DSB repair; and the nuclease meiotic recombination 11 (MRE11), also in DSB repair [
84,
85].
2.1. PARP1 Zn Fingers Bind a DNA Break
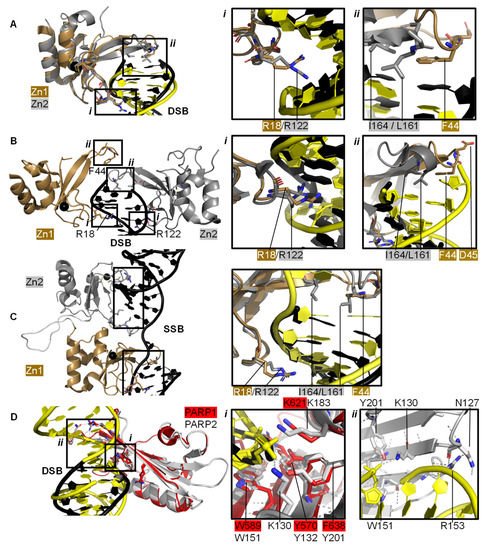
Individual PARP1 domains Zn finger 1 (Zn1) or Zn finger 2 (Zn2) can both bind to DNA with a Cα root mean square deviation (RMSD) of 0.96 Å, when a double-strand DNA molecule representing one end of a DSB is used as a model for DNA damage (PDBs 3ODA, 3ODC, A [
86]). Ali and colleagues [
87] solved the crystal structure of a protein construct containing both Zn1 and Zn2 connected by their linker on a DSB model (PDB 4AV1). This shows Zn1 and Zn2 cooperate to recognise the DSB model (B) yet their binding modes are very similar with a Cα RMSD of 0.80 Å. Zn1 and Zn2 contact DNA at two locations in the phosphate backbone grip (Inset
i in A,B) through R18 (Zn1) or R122 (Zn2) and at the base stacking loop (Inset
ii in A,B) via F44 (Zn1) or L161/I164 (Zn2). In the cooperative interaction these base stacking interactions enhance each other. This happens through hydrophobic protein-DNA interactions mediated by L161/I164 from Zn2 capped with F44 from Zn1. This implicates the Zn finger regions of PARP1 in the binding, and subsequent repair, of DSBs.
Figure 2. PARP domains in complex with a DNA DSB model (
A,
B), SSB (
C), or DSB (
D). (
A) Superposed crystal structures of Zn fingers 1 (Zn1, brown, PDB 3ODA [
86]) and 2 (Zn2, silver, PDB 3ODC [
86]), RMSD 0.96 Å, in complex with DSB models shown in black for Zn1 and yellow for Zn2. Inset
i shows the phosphate backbone grip and inset
ii the base stacking loop; (
B) Zn1 (brown) and Zn2 (silver) cooperate DSB model binding (PDB 4AV1 [
87]). Insets show the superposition of Zn1 and Zn2 with an RMSD of 0.80 Å of the boxed regions. In the superposition, Zn1 and Zn2 binding to a DSB model is shown in black and yellow, respectively, where
i shows the Zn finger loop with R18/R122 interacting with the major groove of the DSB model and
ii the base stacking loop with F44/L161/I164; (
C) Zn1 and Zn2 also cooperate SSB binding (PDB 2N8A [
89]). Inset shows the superposition of the boxed regions in Zn1 and Zn2 on a SSB with an RMSD of 0.83 Å, where DNA in yellow binds to Zn1 and DNA in black binds to Zn2; (
D) Superposition of the WGR domains from PARP1 (red, PDB 4DQY [
34]) on a black DSB model and PARP2 (white, PDB 6F5B [
90]) bridging a yellow DSB with a 5′ phosphate group (Cα RMSD of 0.89 Å). Inset
i highlights the superposed WGR PARP1 and PARP2 residues involved in DNA binding. Inset
ii shows how the WGR of PARP2 interacts with the second DSB using R153.
Both Zn finger interactions are sequence-independent as demonstrated in and the nuclear magnetic resonance spectroscopy (NMR) structures of Zn finger domains (from PARP1 in absence of DNA overlaid with the protein-DNA complex structures [
88]). This shows movement of the base stacking loop of Zn1 upon binding to DNA. Further DNA binding studies imply that the phosphate backbone grip of both Zn1 and Zn2 is key to DNA binding and the base stacking loop contributes modest complementary DNA binding ability. However, only Zn1 is essential for PARP1 activation or PARylation. This “activator” difference is attributed to the sequence diversity between Zn1 and Zn2 surrounding the base stacking loop. A D45A mutation in Zn1, which does not have an equivalent residue in Zn2 in the structure-based sequence alignment, abolishes inter-domain communication within PARP1 leading to PARylation, but this mutation does not prevent DNA binding. This is explained by the fact that D45 in Zn1 points away from the DNA-binding interface and so D45 in Zn1 is essential for inter-domain activation of PARP1 (B, inset
ii). Furthermore, an equivalent residue is absent in Zn2, which explains why Zn2 cannot activate PARP1 on its own [
86].
Eustermann et al. (2011) showed that Zn1 and Zn2 can both bind SSBs in structures obtained by NMR [
88]. The double domain construct Zn1-Zn2 with a flexible 15 amino acid linker binds the SSB model mainly using its Zn2 in a similar manner to Zn2 alone, confirming that Zn2 binds a SSB in preference to Zn1 [
88]. The structural rationale for this was explained by the solution structure of Zn1-Zn2 on a SSB (C) [
89]. Zn2, the domain with a higher binding affinity for DNA, first senses the DNA SSB and interacts with the more accessible 3’ site. This poises Zn1 for DNA binding to the more cryptic 5′ site thereby twisting the DNA; a binding mode only accessible for SSBs. Zn1 and Zn2 also mediate cooperative binding through a hydrophobic interdomain interface (C) [
89]. Remarkably, the key residues for interactions with both SSBs and DSBs are conserved and located in the phosphate backbone grip and base stacking loop. This is a testament to the flexibility of Zn finger domains in PARP1 in binding to different DNA break architectures.
The crystal structure of Zn finger 3 of PARP1 (Zn3, PDB 2RIQ) revealed a Zn finger fold different from Zn1/Zn2 [
22]. Zn3 uses its N-terminal α-helical region to interact with the minor groove of DSBs [
34]. Destabilisation of Zn3 (by the introduction of negative charges in the Zn finger domain structure) resulted in a full-length PARP1 mutant able to bind DNA. However, this was unable to undergo DNA-dependent activation [
91] suggesting that a function of Zn3 is to mediate interdomain contacts upon PARP1 activation by DNA binding. This is confirmed by detailed studies into the order of DNA interaction and PARP1 activation [
34,
89], which describe how DNA binding initiates cooperative interdomain interactions between Zn3 and WGR that support destabilisation of the HD.
The 3.25 Å structure showing the minimal combination of PARP1 domains essential for PARP1 activation on a DSB model comprises Zn1, Zn3, WGR, and the HD-CAT domains [
34], even though Zn2 can bind DSB models in an analogous fashion to Zn1 (PDB 4DQY, B). Here, Zn1 and Zn3 make adjacent interactions to the ribose phosphate backbone of a DSB. They interact with the WGR domain central to PARP1, which binds the 5′ terminus of the DSB via a range of aromatic and positively charged residues in the central β-sheet and Lys600 in its α-helix, and mediates contacts to the CAT domain (B, inset
ii) [
34]. The full-length structure of PARP1 on a DSB remains elusive.
2.2. Recognition and Binding of Other DNA Breaks via PARP Domains
PARP2 and PARP3 lack the Zn finger domains yet still recognise specific DNA breaks featuring 5′ phosphate groups and subsequently initiating a DDR [
76,
90,
92,
93]. Here, the interaction with DNA is mediated by the WGR domain that is also present in PARP1 (D). The structure of PARP2 shows similar binding modes for DNA with (PDB 6F5B) and without (PDB 6F1K) 5′ phosphate group [
90] despite preferentially binding damaged DNA featuring a 5′ phosphate group [
76]. The key interaction in the PARP2-5′ P-DNA appears to be Y201, which coordinates a hydrogen bond to the 5′ phosphate group in PDB 6F5B (D, inset
i), together with the lysine residues K130 and K183. In both structures the PARP2 WGR bridges a DSB (D, inset
ii)—a feasible model of the physiological interaction of the DNA-binding recruitment domain of DNA damage sensor PARP2.
In addition, the N-terminus of PARP2 bears DNA-binding activity and assists in the activation of PARP2 on SSBs [
42]. This region also activates PARP2 in the presence of single-strand RNA molecules, but not double-strand DNA molecules [
36].
2.3. The PARP Paradox: Finding a Needle in a Haystack
For effective DNA damage sensing, PARP molecules need to swiftly locate damaged DNA within a highly crowded nuclear environment containing lots of intact DNA: the estimated DNA concentration in the nucleus is 100 mg/mL [
28,
29]. A contributing factor to this is the high nuclear concentration of PARP molecules and in particular PARP1 [
29,
94], estimated to 7–200 μM from 2 × 10
5–10
6 copies of PARP1 per nucleus [
29,
94], a cell volume between 100 and 10,000 μm
3 [
95] and a ratio of nuclear-to-cell volume of 0.08 [
96]. These PARP molecules have a very high binding affinity (low binding constant, K
D) for damaged DNA, ranging from 3 to 62 nM depending on the PARP molecule and DNA damage site [
97,
98], as measured by Sukhanova et al. (2019) by atomic force microscopy (AFM). In this study, competitive binding was revealed between different members of the PARP family for similar binding sites on DNA. PARP1 has the highest affinity for nick sites, followed by nucleotide break sites and had the lowest affinity for abasic/apyrimidinic (AP) sites. In contrast, PARP2 had similar affinity for nicked and AP sites and the lowest affinity for nucleotide break sites [
98]. This confers an intrinsic difference in DNA binding mechanism, as might be expected with PARP2 lacking DNA-binding Zn domains. AFM binding experiments combined with protein volume measurements provided some insight into the observed oligomeric state of PARP molecules on damaged DNA. PARP1 was mostly monomeric on intact, AP sites and gaps with minute dimer formation on nicked sites, while PARP2 preferentially dimerised on gaps and nicked sites and was monomeric on intact and AP sites [
98]. Studies are emerging on the molecular activation mechanism of PARP2 on DSBs.
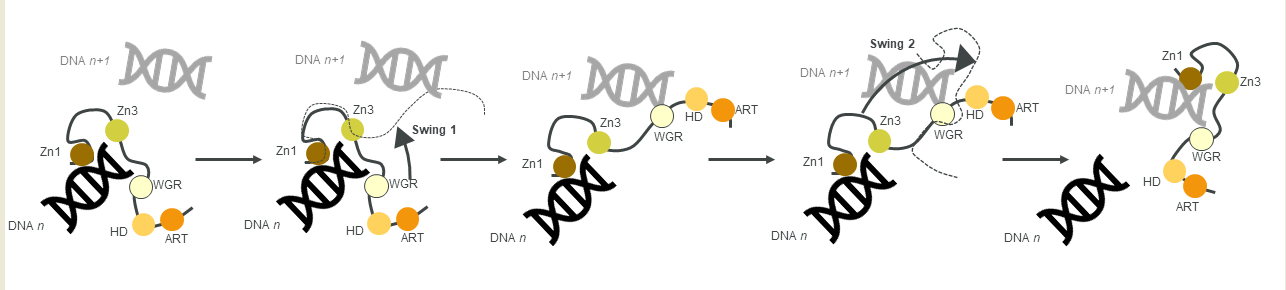
Figure 3. A schematic of the monkey bar mechanism, showing how PARP1 recognises DNA damage.
Another contribution to swift DNA damage sensing comes from the way PARP1 travels along intact DNA—termed the monkey bar mechanism (
Figure 3) [
99]. This intersegment transfer mode is estimated to enhance the ability of PARP1 to sense DNA damage threefold, compared to diffusion alone [
100] (from stopped-flow experiments of PARP1 WT and a W589A mutant unable to use the monkey bar mechanism) [
99,
100]. Moreover, Rudolph et al. (2018) showed that since the association of PARP1 with DNA is faster than diffusion, PARP1 would struggle to release undamaged DNA once bound [
99]. Instead, PARP1 makes good use of excess DNA in the nucleus by dissociating the lower-affinity DNA binding domain WGR from the originally bound DNA-molecule
n. Due to the high DNA concentration in the nucleus, the WGR domain will bind to another DNA-molecule
n + 1. This WGR-DNA interaction is primarily mediated through W589 (D, inset
i), which stacks against the ribose sugar of the 5′ strand of DNA. When the stronger DNA-binding Zn finger domains dissociate from DNA
n they will rapidly bind DNA
n+1. This transfer enables PARP1 to quickly move between DNA molecules, most of which are undamaged until it finds a DNA break, where the Zn domains make specific interactions with the DNA break [
31]. After a conformational change within PARP1, which is most likely pinpointed to the “closure” of the WGR onto DNA that happens on a slower timescale than binding of the Zn fingers to DNA [
100], the HD domain gets destabilised and mediates allosteric activation of the catalytic domain of PARP1. This in turn strengthens the affinity of the Zn finger domains for the DNA break [
32]. In summary, the tailored affinity of different members of the PARP family for different DNA breaks, swinging between high- and lower-affinity DNA-binding domains, and the sheer number of PARP molecules in the nucleus, are the clue towards finding the DNA strand break needle in the DNA haystack called the nucleus.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22105112