Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Hypertension is a medical condition where the blood pressure in the arteries is elevated. However, even if one does not exhibit symptoms, long-term high blood pressure is the major risk factor for numerous pathologies, such as stroke or heart failure. Angiotensin II (AII) receptor blockers are a class of molecules that act in the Renin Angiotensin System (RAS) through binding to the AT1 receptor (Angiotensin II receptor type 1, AT1R) and preventing its activity. AT1R is a G-protein-coupled receptor that is responsible for AII pathophysiological actions.
- rational design
1. Synthetic Routes of Losartan
One common structural characteristic of the commercially AT1R antagonists is that they contain an acidic group such as carboxylate or tetrazole or 5-oxo-1,2,4-oxadiazole ring. Tetrazole and 5-oxo-1,2,4-oxadiazole ring groups are prepared from nitrile group and are bioisosteric. They also retain a more favorable pharmacological effect, pharmacokinetic profile and they are metabolically more stable compared to carboxylate group. The replacement of a functional group by another that modifies beneficially the pharmaceutical profile of a compound is a fundamental medicinal chemistry strategy known as isosteric or bioisosteric replacement [21,22,23,24,25].
It has been claimed that the substitution of the tetrazole ring in candesartan by the structurally identical 5-oxo-1,2,4-oxadiazole ring on azilsartan has led to unique pharmacological properties [26].
In an efficient and green synthetic route to losartan, Shangxia et al. [27] in the last step converted the nitrile derivative to tetrazole ring. Their synthetic methodology is devoid of hazardous chemicals like hydrogen chloride and ammonia, making the process convenient and eco-friendly (Scheme 1, [27]).
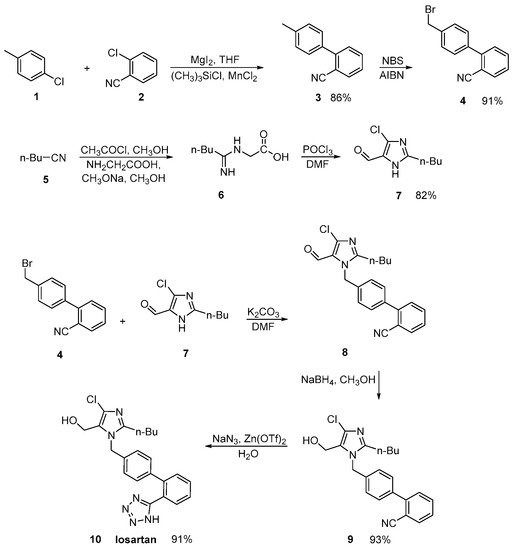
Scheme 1. An efficient synthesis of losartan. The last step resulted in the formation of the tetrazole ring [27].
In a US patent, Reddy A.V. provided the same conversion using a similar reaction (Scheme 2, [28]). The three-step synthetic process uses commercially available 4′-(bromomethyl)-2-cyanobiphenyl (1) as a starting material. The main drawback of this synthesis is the use of excessive hazardous organotin reagent in the last step.
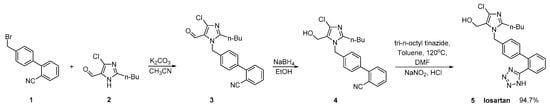
Scheme 2. A patented synthetic route of losartan in which in the last step taking place the conversion of nitrile to tetrazole ring [28].
2. Synthetic Routes for Valsartan
Valsartan was approved from the FDA and released to the US market at 1996 under the name of Diovan. Similar synthetic reagents are described by Ghosh et al. who synthesized valsartan that also bears a tetrazole ring (Scheme 3, [29]). In his work, author described three previous synthetic pathways A–C, which all have the same drawback; the use of expensive boronic acid substrates and palladium catalyst in the cross-coupling step. In a refined approach, inexpensive and commercially available o-anisic acid 10 has been used as the starting material.
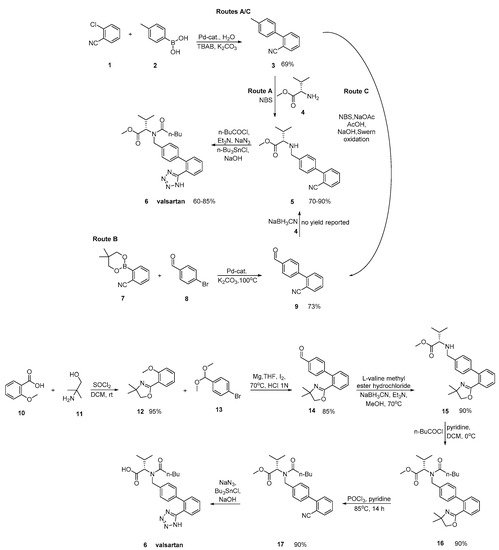
Scheme 3. Synthesis of valsartan through cyclisation of nitrile derivatives 5 and 17 [29].
Zhang et al. (Scheme 4, [30]), Harel et al. (Scheme 5, [31]) and Kankan et al. (Scheme 6, [32]) used nitrile derivatives for cyclization in valsartan.
Zhang emphasized in the ortho-metalation of p-bromotoluene 4 that produces a boronic acid intermediate 10 which was subjected to palladium-catalyzed Suzuki coupling. Implementation of valsartan synthesis using Suzuki reaction is the main advantage of this methodology. The saponification of the methyl ester 11, was realized in a convenient and economical manner and it is more suitable for industrial production. Overall, the synthesis is characterized by minimum use of expensive and hazardous metals and increased efficiency (Scheme 4).
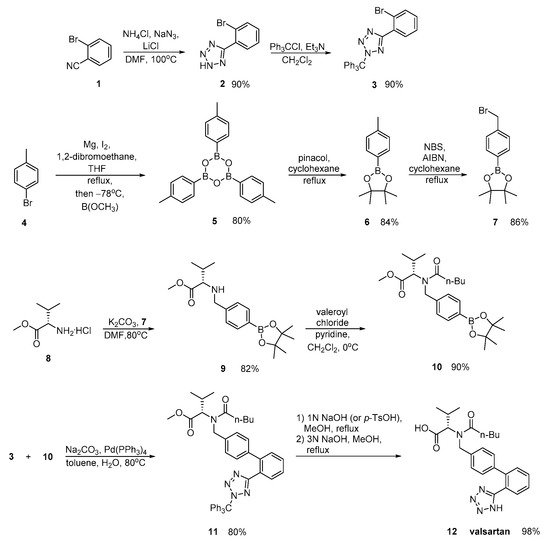
Scheme 4. Synthesis of valsartan through nitrile derivative 1 [30].
In a patent by Harel and Rukhman, a synthetic process for valsartan is incorporated which is substantially free of its isoleucine analogue. Impurities is a major issue in drug synthesis; therefore, their elimination is the focus of this synthetic methodology (Scheme 5).
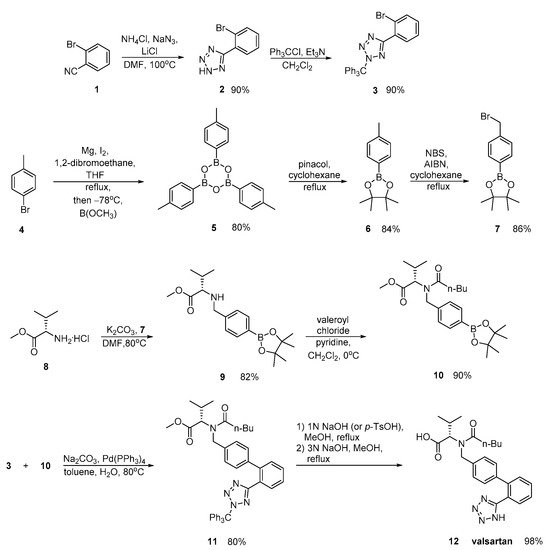
Scheme 5. Synthesis of valsartan through nitrile derivative 7 [31].
In a later patent by Kankan et al., an alternative synthesis was described (Scheme 6). The main disadvantages of this synthesis are: the use of toxic tributyl azide, the formation of explosive hydrogen azide, the oily nature of the intermediates which require repeated step of crystallization making the process laborious and time consuming for industrial scale.
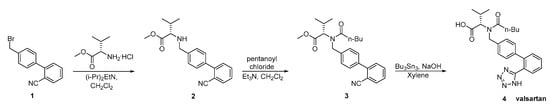
Scheme 6. Synthesis of valsartan through nitrile derivative 3 [32].
3. Synthetic Routes of Irbesartan
Irbesartan was approved from the FDA and released to the US market at 1996 under the name of Avapro. A similar strategy was used for the synthesis of irbesartan (Scheme 7 and Scheme 8, [33,34,35]). In particular, irbesartan was synthesized through dehydration and tetrazole formation in one step from a substituted cyclopentane derivative 6 with tributyltin chloride and sodium azide. The purity of the compound was 99.85% as confirmed by HPLC analysis. Minimization of the impurities was the mail focus of this synthetic methodology (Scheme 7, [33,34]).
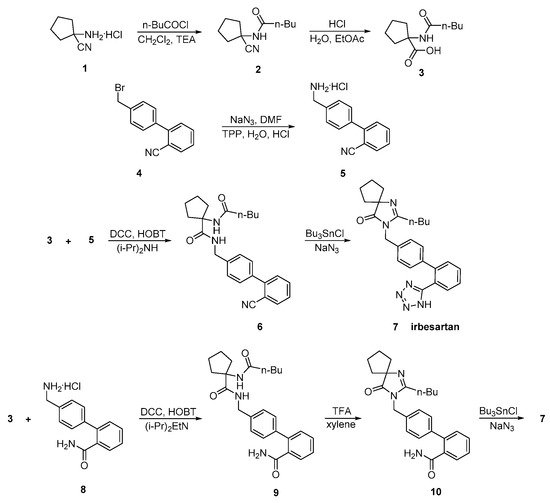
In an elegant approach, irbesartan was synthesized through a new catalytic system based on palladium-amido-N-heterocyclic carbenes for Suzuki–Miyaura coupling reactions of heteroaryl bromides. In this synthetic method, the biaryl structure was formed using a very low catalytic load making the procedure industrial friendly. Thus, the use of the industry-advantageous Suzuki reaction is the hallmark of this synthetic methodology (Scheme 8, [35]).
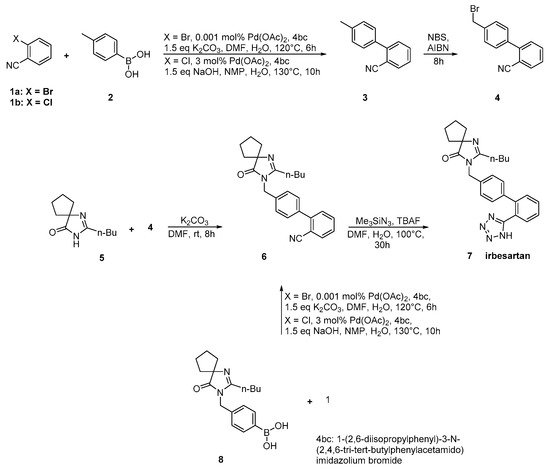
Scheme 8. Synthesis of irbesartan through nitrile derivative 6 by Kumar, A.S et al. [35].
4. Synthetic Routes of Azilsartan
Azilsartan was approved from the FDA and released to the US market at 2011 under the name of Edarbi. Nitriles are also useful for the synthesis of 1,2,4-oxadiazol ring of azilsartan (Scheme 9 and Scheme 10, [36,37]). The method described in Scheme 10, [36] is superior to previous published (Scheme 9, [37]) because it is very efficient (90% yield) and does control the impurities (99.93% HPLC purity).
It must be pointed out that impurities in the synthesis of AT1R antagonists is an important issue. It is suffice to say that recently Princeton Pharmaceuticals recalled numerous irbesartan tablets and seven lots of irbesartan hydrochlorothiazide (HCTZ) because after testing revealed the drugs contained trace amounts of a carcinogen [38,39].
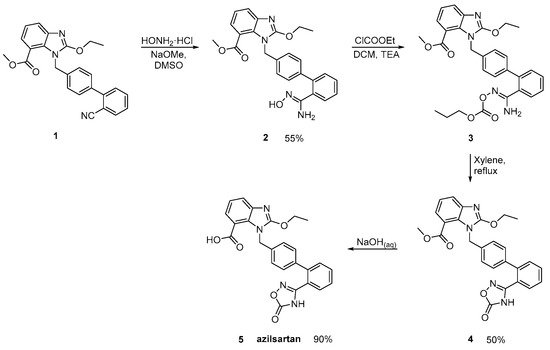
Scheme 9. Synthesis of azilsartan. The nitrile derivative 1 is cyclized to final product 5 (azilsartan) [37].
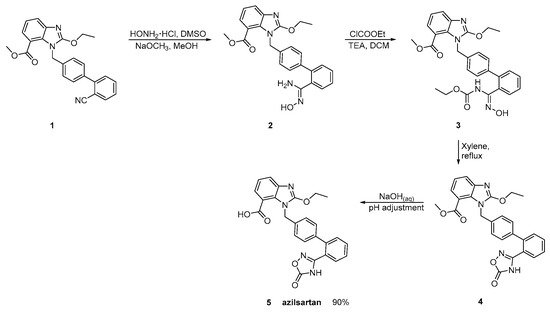
Scheme 10. Synthesis of azilsartan. Improved synthesis of the nitrile derivative 1 is cyclized to final product 5 (azilsartan) [36].
5. Synthetic Routes of Telmisartan
Telmisartan was approved from the FDA at 1998 and released to the US market at 2000 under the name of Micardis. Telmisartan is assembled by two benzimidazole subunits and a biphenyl-2-carboxylic acid segment. A nitrile group can also be used to produce the carboxylate group of telmisartan through hydrolysis. The benzimidazole moiety can be created through an improved, copper-catalyzed, cyclisation of o-haloarylamidines obtained from o-haloarylamines. The total synthesis started from the commercially available 4-Nitro-m-toluic acid and it was achieved in seven steps with an overall yield of 54%. The advantage of this synthesis, relatively to the previous reported, is the absence of the use of possibly harmful HNO3/H2SO4 for nitration and polyphosphoric acid (PPA) for cyclisation (Scheme 11, [40]).
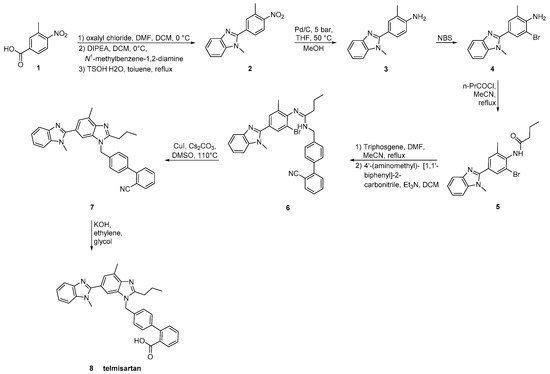
Scheme 11. Synthesis of telmisartan by Zhang, J et al. The nitrile derivative 7 in the last step is converted to carboxylate analog 8 [40].
Telmisartan has also been prepared through different methodologies where the dimer benzimidazole rings are incorporated (Scheme 12, Scheme 13, Scheme 14, Scheme 15 and Scheme 16, [14,41,42,43,44]).
The original synthesis of telmisartan was developed by Ries et al. in 1993 (Scheme 12, [14]), beginning with the stepwise construction of the central benzimidazole ring from 4-amino-m-toluic acid methyl ester 1. Saponification of the resulting substituted benzimidazole 2 was followed by condensation with N-methyl-1,2-phenylenediamine using polyphosphoric acid at elevated temperature (150 °C) to afford the functionalized dibenzimidazole 3. Alkylation of the latter with 4′-(bromomethyl)-2-biphenylcarboxylic acid tert-butyl ester 4 followed by hydrolysis of the resulting ester provided telmisartan in 21% overall yield over eight linear steps. Albeit the relatively low overall yield, the synthetic methodology is attractive because of the reactions’ simplicity and the availability of the cheap reagents.
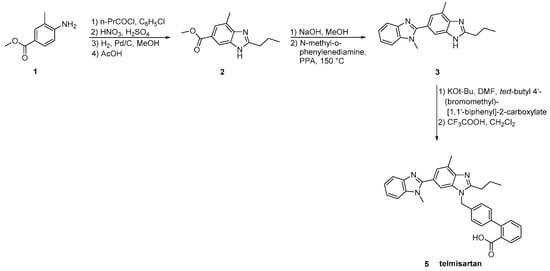
Scheme 12. First reported synthesis of telmisartan by Uwe J. Ries et al. [14].
Goossen et al. (Scheme 13, [41]) introduced the biaryl moiety of telmisartan using a Pd/Cu decarboxylative catalyst system. Construction of the biaryl moiety with the aid of a powerful tool like decarboxylative coupling instead of using appropriate reagents containing it is the characteristic of this synthetic pathway.
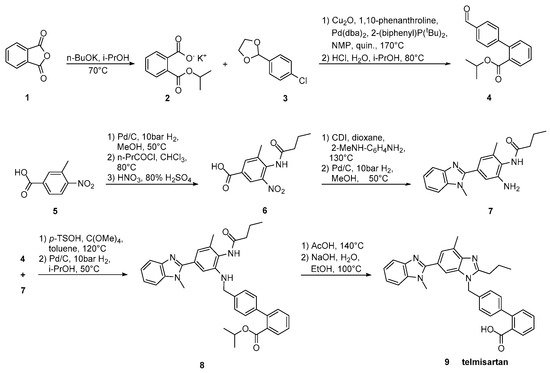
Scheme 13. Synthesis of telmisartan by Lukas J. Goossen et al. via decarboxylative cross-coupling [41].
In the method reported by Kumar et al. (Scheme 14, [44]), the biaryl moiety of telmisartan was elaborated by the directed metalation of an appropriate carboxy protecting group followed by transmetalation with zinc chloride and then transition metal catalyzed cross coupling with aryl bromides. Construction of the biaryl moiety is again the hallmark of this synthesis.
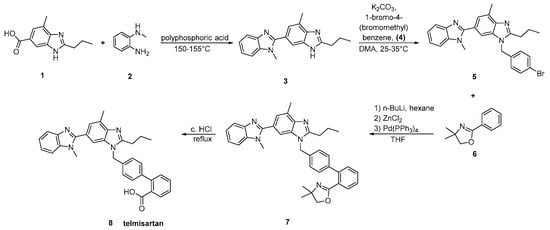
Scheme 14. A modified synthesis of telmisartan by A. Sanjeev Kumar et al. [44].
The key strategy in the synthesis reported by Wang et al. (Scheme 15, [42]) was the construction of bis-benzimidazole 7 by reductive cyclization of o-nitroaniline 6 with butyl aldehyde. The overall process is operational simple, with low production cost.
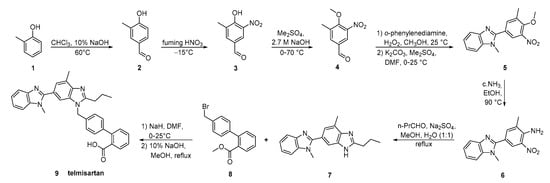
Scheme 15. A highly practical and cost-effective synthesis of telmisartan by Ping Wang et al. [42].
In the synthesis reported by Martin et al. (Scheme 16, [43]), two differentially substituted benzimidazole derivatives and a biphenyl-2-carboxylic acid synthon were assembled via a Suzuki cross-coupling reaction, providing telmisartan in 72% overall yield. High overall yielding synthetic methodology makes this procedure attractive.
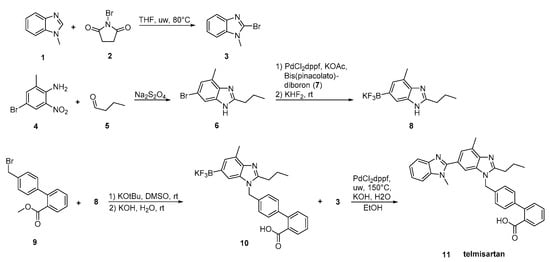
Scheme 16. Total synthesis of telmisartan by Alex D. Martin et al. via a Suzuki cross-coupling reaction between two functionalized benzimidazoles [43].
6. Synthetic Routes of Eprosartan
Eprosartan was approved from the FDA at 2001 and released to the US market at 2003 under the name of Teveten. Eprosartan is one of the AT1R antagonists that does not contain a biphenyl tetrazole ring. It contains two heterocycles, the imidazole and the unique thiophene. Below are reported synthetic routes used to develop it (Scheme 17, Scheme 18, Scheme 19 and Scheme 20, [45,46,47,48,49]).
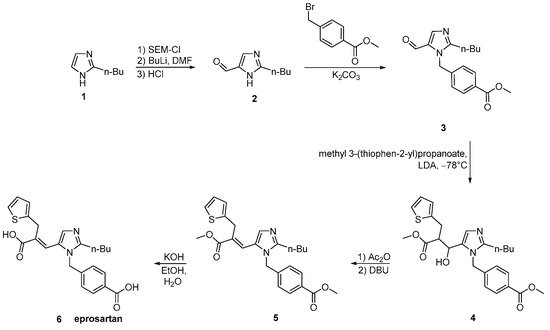
Scheme 17. Synthesis of eprosartan by Finkelstein et al. [49].
The process described by Ramakrishnan et al. (Scheme 18, [46]) comprises condensation of valerimidine methyl ester with dihydroxyacetone to give a diacetate which was treated with α-carboxymethylbenzyl alcohol in the presence of triflic acid to give 2-n-butyl-5-acetoxymethyll-(4-carboxyphenyl)-methyl-lH-imidazole. Thus obtained compound on further oxidization with manganese dioxide and thereafter condensation with methyl-3-(3-(2-theinyl)-propionate in the presence of n-butyl lithium at −78 °C gives an ester which is hydrolyzed to give eprosartan. Eprosartan is further converted to its desired salt form.
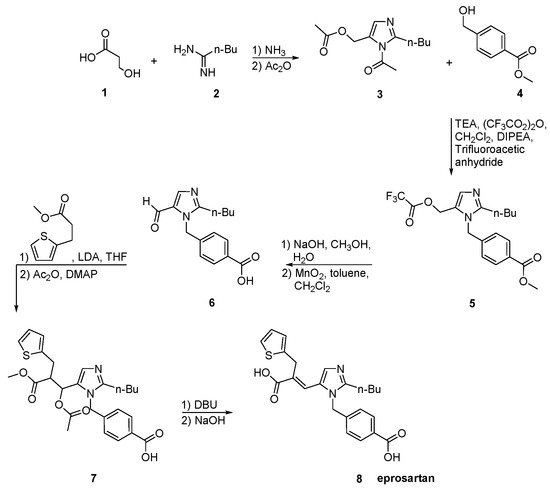
Scheme 18. Synthesis of eprosartan by Ramakrishnan et al. [46].
Eprosartan synthesis was reported by Matsuoka et al. to proceed in three stages (Scheme 19, [45]). These stages are: the regioselective protection of 2-n-butyl-4-formylimidazole 1, followed by reaction with (2-thienyimethyl)-propanedioic acid, mono-alkyl ester and 4-(bromomethyl)benzoate. The efficiency of this synthetic sequence and the quality and yield of eprosartan are particularly important.
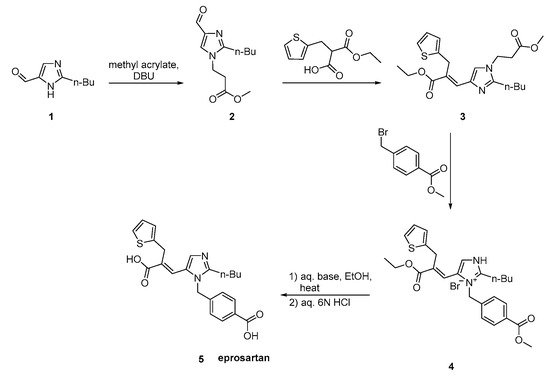
Scheme 19. Synthesis of eprosartan by Matsuoka et al. [45].
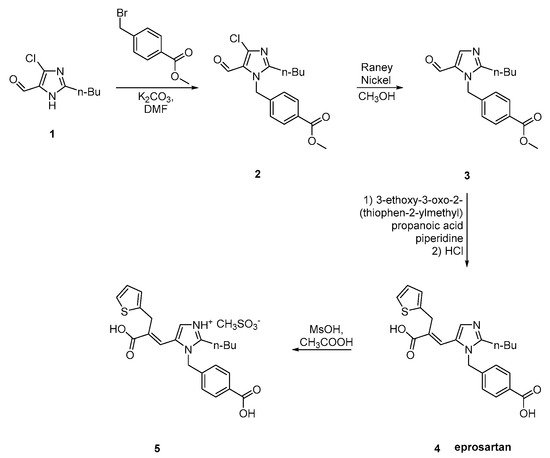
7. Synthetic Routes of Olmesartan Medoxomil & Candesartan Cilexetil
Olmesartan and candesartan are given as pro-drugs olmesartan medoxomil and candesartan cilexetil. Olmesartan was approved from the FDA and released to the US market at 2002 under the name of Benicar while candesartan was approved at 1998 and released to the US market at 1999 under the name of Atacand. Synthetic routes are provided in the following schemes (Scheme 21, Scheme 22, Scheme 23 and Scheme 24, [50,51,52,53]).
Candesartan was synthesized using an efficient protocol for C–H arylation by a catalytic system involving [RuCl2(p-cymene)]2 and triphenylphosphine (Scheme 21, [50]).
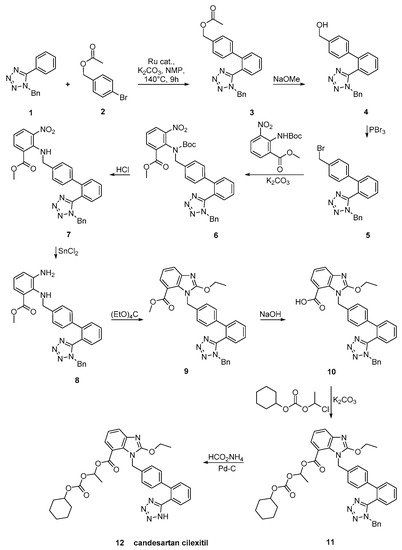
Scheme 21. A practical synthesis of prodrug candesartan cilexitil by Masahiko Seki et al. [50].
By careful monitoring of the reaction conditions, Babu et al., described an olmesartan medoxomil synthesis, where impurities were minimized (Scheme 22, [51]).
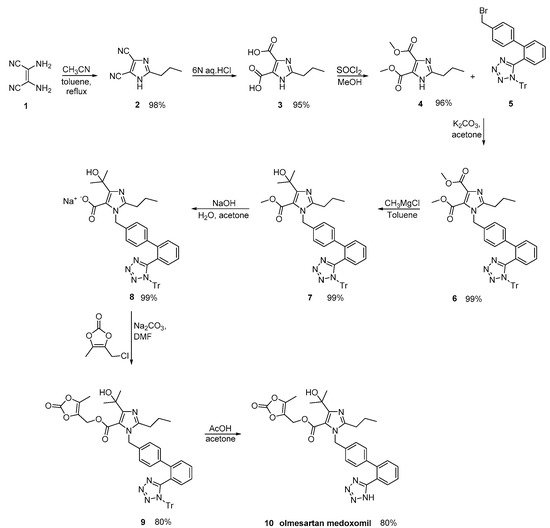
Scheme 22. Synthesis of the prodrug olmesartan medoxomil by Babu et al. [51].
Venkanna et al. identified the impurities resulting from the olmesartan medoxomil synthetic process (Scheme 23, [52]).
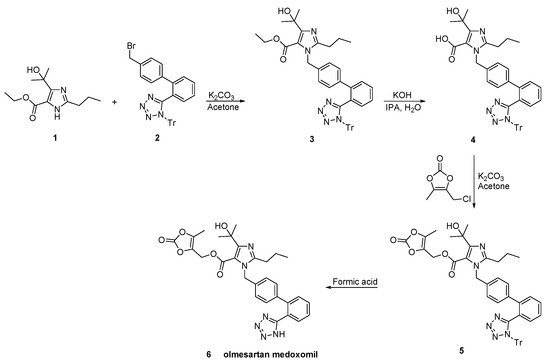
Scheme 23. Synthesis of the prodrug olmesartan medoxomil by Venkanna et al. [52].
In a patent filled by Toplak Casar [53], two variations of an one-pot trityl olmesartan medoxomil synthesis have been described (Scheme 24 and Scheme 25). In this methodology, the intermediates are not isolated in each step and by using the same solvent and the same base, no material is removed or exchanged. This feature renders this methodology particularly attractive.
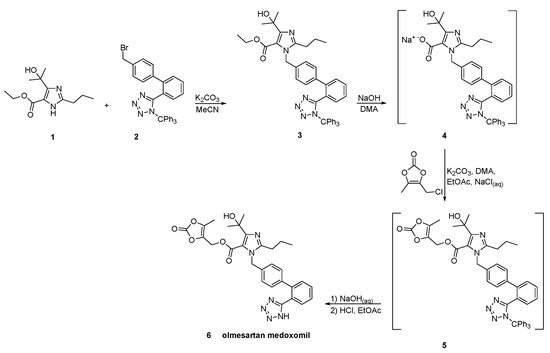
Scheme 24. First variation of one-pot synthesis of the prodrug olmesartan medoxomil [53].
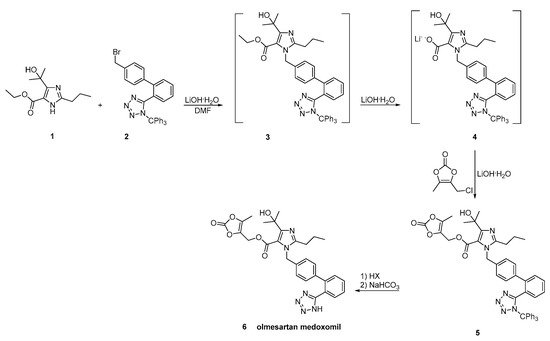
Scheme 25. Second variation of one-pot synthesis of the prodrug olmesartan medoxomil [53].
This entry is adapted from the peer-reviewed paper 10.3390/molecules26102927
This entry is offline, you can click here to edit this entry!