Pulmonary hypertension is defined as a group of diseases characterized by a progressive increase in pulmonary vascular resistance (PVR), which leads to right ventricular failure and premature death. There are multiple clinical manifestations that can be grouped into five different types. Pulmonary artery remodeling is a common feature in pulmonary hypertension (PH) characterized by endothelial dysfunction and smooth muscle pulmonary artery cell proliferation. The current treatments for PH are limited to vasodilatory agents that do not stop the progression of the disease. Therefore, there is a need for new agents that inhibit pulmonary artery remodeling targeting the main genetic, molecular, and cellular processes involved in PH. Chronic inflammation contributes to pulmonary artery remodeling and PH, among other vascular disorders, and many inflammatory mediators signal through the JAK/STAT pathway. Recent evidence indicates that the JAK/STAT pathway is overactivated in the pulmonary arteries of patients with PH of different types. In addition, different profibrotic cytokines such as IL-6, IL-13, and IL-11 and growth factors such as PDGF, VEGF, and TGFβ1 are activators of the JAK/STAT pathway and inducers of pulmonary remodeling, thus participating in the development of PH. The understanding of the participation and modulation of the JAK/STAT pathway in PH could be an attractive strategy for developing future treatments.
- pulmonary hypertension (PH)
- Janus kinase 2 (JAK2)
- signal transducer and activator of transcription 3 (STAT3)
1. Introduction
2. JAK/STAT Pathway and Cellular and Molecular Dysregulation in Pulmonary Hypertension
2.1. JAK/STAT Pathway and Vascular Remodeling
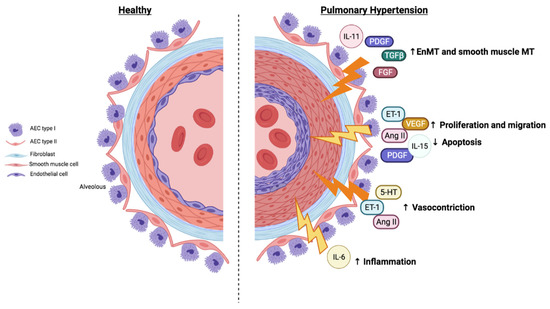
2.2. JAK/STAT Pathway, Proliferation, and Resistance to Apoptosis
2.3. JAK/STAT Pathway, Migration, and Angiogenesis
2.4. Imbalance in Vasoactive Mediators: Vasoconstriction
2.5. JAK/STAT Pathway and Inflammation Associated with Pulmonary Hypertension
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094980
References
- Kondo, T.; Okumura, N.; Adachi, S.; Murohara, T. Pulmonary Hypertension: Diagnosis, Management, and Treatment. Nagoya J. Med. Sci. 2019, 81, 19–30.
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913.
- Zuo, L.; Rose, B.A.; Roberts, W.J.; He, F.; Banes-Berceli, A.K. Molecular Characterization of Reactive Oxygen Species in Systemic and Pulmonary Hypertension. Am. J. Hypertens. 2014, 27, 643–650.
- Liu, C.; Arnold, R.; Henriques, G.; Djabali, K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells 2019, 8, 1276.
- Zhang, H.; Watanabe, R.; Berry, G.J.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation 2018, 137, 1934–1948.
- Paulin, R.; Meloche, J.; Bonnet, S. STAT3 signaling in pulmonary arterial hypertension. JAK-STAT 2012, 1, 223–233.
- Milara, J.; Ballester, B.; Morell, A.; Ortiz, J.L.; Escrivá, J.; Fernández, E.; Perez-Vizcaino, F.; Cogolludo, A.; Pastor, E.; Artigues, E.; et al. JAK2 mediates lung fibrosis, pulmonary vascular remodelling and hypertension in idiopathic pulmonary fibrosis: An experimental study. Thorax 2018, 73, 519–529.
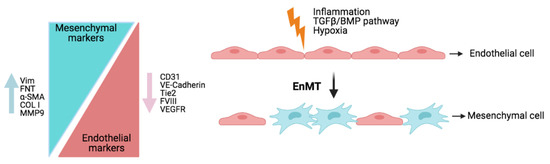
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018.
- Roger, I.; Milara, J.; Montero, P.; Ribera, P.; Cortijo, J. IL-11 promotes pulmonary vascular remodeling and lung fibrosis through the activation of endothelial to mesenchymal transition. Eur. Respir. J. 2020, 56, 3378.
- Leopold, J.A.; Maron, B.A. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2016, 17, 761.
- Hopper, R.K.; Moonen, J.-R.A.J.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 2016, 133, 1783–1794.
- Suzuki, T.; Carrier, E.J.; Talati, M.H.; Rathinasabapathy, A.; Chen, X.; Nishimura, R.; Tada, Y.; Tatsumi, K.; West, J. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L118–L126.
- Paulin, R.; Courboulin, A.; Meloche, J.; Mainguy, V.; de la Roque, E.D.; Saksouk, N.; Côté, J.; Provencher, S.; Sussman, M.A.; Bonnet, S. Signal Transducers and Activators of Transcription-3/Pim1 Axis Plays a Critical Role in the Pathogenesis of Human Pulmonary Arterial Hypertension. Circulation 2011, 123, 1205–1215.
- Shibata, R.; Kai, H.; Seki, Y.; Kato, S.; Wada, Y.; Hanakawa, Y.; Hashimoto, K.; Yoshimura, A.; Imaizumi, T. Inhibition of STAT3 prevents neointima formation by inhibiting proliferation and promoting apoptosis of neointimal smooth muscle cells. Hum. Gene Ther. 2003, 14, 601–610.
- Cibull, T.L.; Jones, T.D.; Li, L.; Eble, J.N.; Baldridge, L.A.; Malott, S.R.; Luo, Y.; Cheng, L. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J. Clin. Pathol. 2006, 59, 285–288.
- Chiang, W.-F.; Yen, C.-Y.; Lin, C.-N.; Liaw, G.-A.; Chiu, C.-T.; Hsia, Y.-J.; Liu, S.-Y. Up-regulation of a serine–threonine kinase proto-oncogene Pim-1 in oral squamous cell carcinoma. Int. J. Oral Maxillofac. Surg. 2006, 35, 740–745.
- Beier, U.H.; Weise, J.B.; Laudien, M.; Sauerwein, H.; Görögh, T. Overexpression of Pim-1 in head and neck squamous cell carcinomas. Int. J. Oncol. 2007, 30, 1381–1387.
- Masri, F.A.; Xu, W.; Comhair, S.A.A.; Asosingh, K.; Koo, M.; Vasanji, A.; Drazba, J.; Anand-Apte, B.; Erzurum, S.C. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L548–L554.
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1798–H1809.
- Courboulin, A.; Paulin, R.; Giguère, N.J.; Saksouk, N.; Perreault, T.; Meloche, J.; Paquet, E.R.; Biardel, S.; Provencher, S.; Côté, J.; et al. Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 2011, 208, 535–548.
- Udjus, C.; Cero, F.T.; Halvorsen, B.; Behmen, D.; Carlson, C.R.; Bendiksen, B.A.; Espe, E.K.S.; Sjaastad, I.; Løberg, E.M.; Yndestad, A.; et al. Caspase-1 induces smooth muscle cell growth in hypoxia-induced pulmonary hypertension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L999–L1012.
- Yahata, Y.; Shirakata, Y.; Tokumaru, S.; Yamasaki, K.; Sayama, K.; Hanakawa, Y.; Detmar, M.; Hashimoto, K. Nuclear translocation of phosphorylated STAT3 is essential for vascular endothelial growth factor-induced human dermal microvascular endothelial cell migration and tube formation. J. Biol. Chem. 2003, 278, 40026–40031.
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374.
- Cool, C.D.; Stewart, J.S.; Werahera, P.; Miller, G.J.; Williams, R.L.; Voelkel, N.F.; Tuder, R.M. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am. J. Pathol. 1999, 155, 411–419.
- Manish, M.; Markus, R.; Peter, K.; Simone, H.; Eva, D.; Parag, G.; Anne-Christin, S.; Theo, S.R.; Ardeschir, G.H.; Grazyna, K.; et al. Hypoxia-Dependent Regulation of Nonphagocytic NADPH Oxidase Subunit NOX4 in the Pulmonary Vasculature. Circ. Res. 2007, 101, 258–267.
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.K.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L2–L10.
- Frazziano, G.; Champion, H.C.; Pagano, P.J. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H2166–H2177.
- Dennis, K.E.; Aschner, J.L.; Milatovic, D.; Schmidt, J.W.; Aschner, M.; Kaplowitz, M.R.; Zhang, Y.; Fike, C.D. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 297, L596–L607.
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med. 2012, 52, 1970–1986.
- Aggarwal, S.; Gross, C.M.; Sharma, S.; Fineman, J.R.; Black, S.M. Reactive oxygen species in pulmonary vascular remodeling. Compr. Physiol. 2013, 3, 1011–1034.
- Jin, H.; Liu, M.; Zhang, X.; Pan, J.; Han, J.; Wang, Y.; Lei, H.; Ding, Y.; Yuan, Y. Grape seed procyanidin extract attenuates hypoxic pulmonary hypertension by inhibiting oxidative stress and pulmonary arterial smooth muscle cells proliferation. J. Nutr. Biochem. 2016, 36, 81–88.
- Freund-Michel, V.; Guibert, C.; Dubois, M.; Courtois, A.; Marthan, R.; Savineau, J.-P.; Muller, B. Reactive oxygen species as therapeutic targets in pulmonary hypertension. Ther. Adv. Respir. Dis. 2013, 7, 175–200.
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2013, 20, 2794–2814.
- Ding, L.; Chapman, A.; Boyd, R.; Wang, H.D. ERK activation contributes to regulation of spontaneous contractile tone via superoxide anion in isolated rat aorta of angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2997–H3005.
- Nakane, H.; Miller, F.J.; Faraci, F.M.; Toyoda, K.; Heistad, D.D. Gene transfer of endothelial nitric oxide synthase reduces angiotensin II-induced endothelial dysfunction. Hypertens. Dallas Tex 1979 2000, 35, 595–601.
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339.
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertens. Dallas Tex 1979 1998, 32, 488–495.
- Sud, N.; Black, S.M. Endothelin-1 Impairs Nitric Oxide Signaling in Endothelial Cells Through a Protein Kinase Cδ-Dependent Activation of STAT3 and Decreased Endothelial Nitric Oxide Synthase Expression. DNA Cell Biol. 2009, 28, 543–553.
- Bhaskaran, S.; Zaluski, J.; Banes-Berceli, A. Molecular interactions of serotonin (5-HT) and endothelin-1 in vascular smooth muscle cells: In vitro and ex vivo analyses. Am. J. Physiol. Cell Physiol. 2014, 306, C143–C151.
- Wallace, T.A.; Xia, S.-L.; Sayeski, P.P. Jak2 tyrosine kinase prevents angiotensin II-mediated inositol 1,4,5 trisphosphate receptor degradation. Vascul. Pharmacol. 2005, 43, 336–345.
- Rabinovitch, M.; Gamble, W.; Nadas, A.S.; Miettinen, O.S.; Reid, L. Rat pulmonary circulation after chronic hypoxia: Hemodynamic and structural features. Am. J. Physiol. 1979, 236, H818–H827.
- Pugliese, S.C.; Poth, J.M.; Fini, M.A.; Olschewski, A.; El Kasmi, K.C.; Stenmark, K.R. The role of inflammation in hypoxic pulmonary hypertension: From cellular mechanisms to clinical phenotypes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L229–L252.
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175.
- El Chami, H.; Hassoun, P.M. Inflammatory mechanisms in the pathogenesis of pulmonary arterial hypertension. Compr. Physiol. 2011, 1, 1929–1941.
- Price, L.C.; Wort, S.J.; Perros, F.; Dorfmüller, P.; Huertas, A.; Montani, D.; Cohen-Kaminsky, S.; Humbert, M. Inflammation in pulmonary arterial hypertension. Chest 2012, 141, 210–221.
- Kurakula, K.; Smolders, V.F.E.D.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.-J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57.
- Chava, K.R.; Karpurapu, M.; Wang, D.; Bhanoori, M.; Kundumani-Sridharan, V.; Zhang, Q.; Ichiki, T.; Glasgow, W.C.; Rao, G.N. CREB-Mediated IL-6 Expression Is Required for 15(S)-Hydroxyeicosatetraenoic Acid–Induced Vascular Smooth Muscle Cell Migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 809–815.
- Wen, Y.; Gu, J.; Chakrabarti, S.K.; Aylor, K.; Marshall, J.; Takahashi, Y.; Yoshimoto, T.; Nadler, J.L. The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-alpha in macrophages. Endocrinology 2007, 148, 1313–1322.
- Elaine, S.; Holmes Alan, M.; Treacy Carmen, M.; Doughty Natalie, J.; Laura, S.; Machado Rajiv, D.; Trembath Richard, C.; Simon, J.; Lucy, B.; Paul, N.; et al. Elevated Levels of Inflammatory Cytokines Predict Survival in Idiopathic and Familial Pulmonary Arterial Hypertension. Circulation 2010, 122, 920–927.
- Graham, B.B.; Chabon, J.; Kumar, R.; Kolosionek, E.; Gebreab, L.; Debella, E.; Edwards, M.; Diener, K.; Shade, T.; Bifeng, G.; et al. Protective Role of IL-6 in Vascular Remodeling in Schistosoma Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 951–959.