Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell & Tissue Engineering
Interferons (IFNs) are induced by viruses and are the main regulators of the host antiviral response. They balance tissue tolerance and immune resistance against viral challenges.
- type I interferon
- neutrophils
1. Recognition of Viral PAMPs by Neutrophils
Neutrophils are the largest proportion of any cell subset within the innate immune system [1] and have traditionally been thoroughly characterized as an effective component of bacterial pathogen clearance. Research over the past decade has emphasized the expanding role of these innate cells in viral clearance [2,3,4]. Innate immune responses are initiated by recognition of PAMPs by a limited array of specific pattern recognition receptors (PRRs) expressed in and on sentinel cells. Recognition of viral PAMPs by PRRs—expressed by hematopoietic and non-hematopoietic cells of the immune system—results in the activation of intracellular signaling pathways, mediated by several interconnected adaptor proteins. Toll-like receptors (TLRs), which are an important class of PRRs, signal through a range of adaptor proteins. These virus-induced intracellular signaling pathways eventually converge on IFN regulatory factor (IRF)-mediated upregulation of IFNs and IFN-stimulated genes (ISGs) (Figure 1)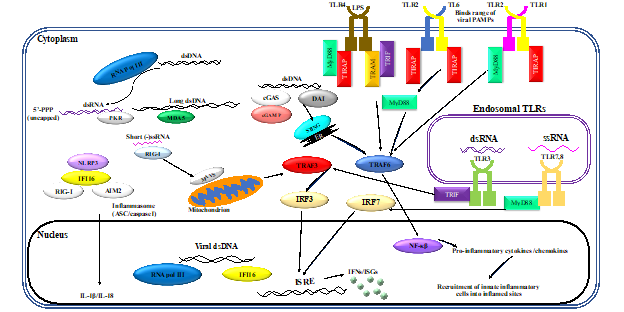
Figure 1. Virus-induced inflammatory responses. Recognition of viral pathogen-associated molecular patterns (PAMPs) by innate cells of the immune system results in inflammatory responses. Activation of innate leukocytes via pattern recognition receptors (PRRs) that recognize viral PAMPs in different cellular compartments gives rise to a number of intracellular signaling cascades, mediated by various interconnected adaptor proteins. This results in interferon regulatory factor (IRF)-mediated upregulation of interferons (IFNs) and interferon-stimulated genes (ISGs), as well as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-mediated induction of inflammatory cytokines and chemokines. Furthermore, sensing of viral PAMPs by NOD-like receptor family pyrin domain containing 3 (NLRP3), retinoic acid-inducible gene I (RIG-I), absent in melanoma 2-like receptors (AIM2), and/or IFN-inducible protein 16 (IFI16) potentiates the formation of inflammasome complexes, which ultimately result in the induction of inflammatory cytokines such as interleukin (IL)-1β and IL-18. Other abbreviations: cGAMP: cyclic guanosine monophosphate–adenosine monophosphate, cGAS: cyclic guanosine monophosphate–adenosine monophosphate synthase, DAI: deoxyribonucleic acid (DNA)-dependent activator of interferon regulatory factors, ds: double-stranded, ER: endoplasmic reticulum, ISRE: interferon-sensitive response element, MAVS: mitochondrial antiviral signaling protein, MDA5: melanoma differentiation-associated protein 5, MyD88: myeloid differentiation primary response 88, NOD: nucleotide-binding oligomerization domain, PKR: protein kinase R, pol: polymerase, RNA: ribonucleic acid, ss: single-stranded, STING: stimulator of interferon genes, TIRAP: Toll/interleukin-1 receptor (TIR) domain-containing adapter protein, TLR: toll-like receptor, TRAF: tumor necrosis factor receptor–associated factor, TRAM: TIR-domain-containing adapter-inducing IFN-β (TRIF)-related adaptor molecule.
Neutrophils express a broad repertoire of PRRs and respond to PRR ligation during viral infection and inflammation. Neutrophils express all TLRs except for TLR3 [5]. Granulocyte-macrophage colony stimulating factor (GM-CSF), which controls different cell functions in inflammation, can promote neutrophil survival and trafficking; it can also upregulate oxidative burst, phagocytosis, and formation of extracellular traps [6], and increase both TLR2 and TLR9 expression in neutrophils [5]. TLR4, which recognizes lipopolysaccharide (LPS), was shown to be required for neutrophil migration to the lungs [7]. Neutrophils frequently travel to the lungs after a range of viral infections, including those caused by respiratory syncytial virus (RSV), highly pathogenic avian influenza virus, influenza A virus (IAV) [1], and vesicular stomatitis virus (VSV) [8]. However, neutrophils are still capable of killing a range of pathogens independent of TLRs [9]. For instance, it has been shown that neutrophil-derived IFN-γ is required for TLR-independent host protection against intracellular pathogens [10].
Studies were conducted on the interactions between viruses and neutrophil TLRs [11]. Neutrophils quickly upregulated TLR2 expression after exposure to IAV [12], and neutrophils treated with IAV increased their ability to phagocytize other pathogens. The single-stranded RNA recognition receptors, TLR7 and TLR8, were also involved in the neutrophil response to IAV [13]. TLR4 signaling in plasmid-transfected neutrophils resulted in expression of IFN-β [14]. Similar production of IFN- β was documented for a wide range of pathogens, including human adenovirus serotype 5. The TLR4 agonist LPS resulted in additional upregulation of IFN-β transcripts [14] and multiple research groups have elucidated a link between TLRs and the production of antiviral interferons. It is unclear if the TLR/type I IFN axis could somehow be modulated in a way to gain an appropriate antiviral immune response while minimizing tissue damage.
2. Regulation of IFN Signaling in Neutrophils
Neutrophils’ ability to produce IFNs in conjunction with recognition of viral PAMPs suggests that they are critical for innate antiviral host defenses. Using a range of stimulatory compounds, researchers demonstrated that messenger RNAs encoding IFN-α, -β, and -γ were constitutively expressed in neutrophils [5]. The presence of type I IFNs reduces the concentration of lipid A, a TLR4 agonist that is required to induce TRIF-dependent genes, demonstrating a link between TLR4 and IFNs [80]. Neutrophils can use helicase recognition to activate a robust antiviral response [81]. The viral double-stranded RNA mimetic poly(I:C) can be recognized by neutrophils, despite them not possessing TLR3. Constitutive expression of MDA5 and RIG-I aids neutrophils in recognizing the viral genetic material and subsequently producing type I IFNs, IFN-responsive genes (IRGs), and immunoregulatory cytokines. These findings were reinforced in experiments using encephalomyocarditis virus in MDA5-deficient mice, which have a reduction in IFN-β production [81].
Mature neutrophils are predominantly responsible for neutrophil-mediated IFN responses, as immature neutrophils do not express IFNARs and have lower IRG expression levels [82]. Immature neutrophils are also incapable of effectively phosphorylating STAT1 and are not primed effectively by IFNs. Likewise, studies of immature neutrophil gene regulation illustrated limited IFN ability to control immature neutrophil proliferation. Although IFNs did not have an effect on immature neutrophils, IFN-α does influence their precursor hematopoietic stem cells by activating dormant cells [83]. In contrast, mature neutrophils express genes to enable them to respond to both type I and II IFNs [82]. IFN-α primes mature neutrophils, enabling them to form neutrophil extracellular traps (NETs) to bind to pathogens (Figure 2). In a positive feedback loop, these traps—which are composed primarily of DNA, high mobility group box protein 1 (HMGB1), and the cathelicidin antimicrobial peptide LL37—subsequently activate pDCs, which in turn produce more IFN-α via DNA binding to TLR9 [84]. Interferon-deficient mice have reduced production of NETs and reactive oxygen species (ROS), while recombinant IFN-β treatment restored NETosis [85]. Controlling this feedback loop may be a method warranting further examination in diseases that are exacerbated by excessive formation of NETs.
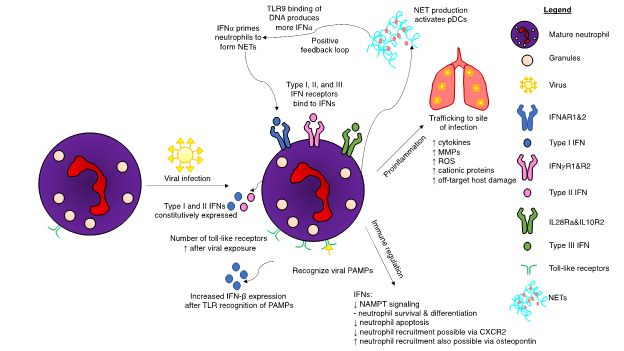 Figure 2. Interferon (IFN)-mediated regulation of neutrophils upon viral infection of a host. Viral infections induce many changes to neutrophil biology. Toll-like receptor (TLR) expression increases after viral exposure, resulting in increased production of type I interferons (IFNs). Neutrophils possess receptors for all three interferon subsets. A positive feedback loop occurs during the production of neutrophil extracellular traps (NETs). IFNα results in NET production, which in turn activates plasmacytoid dendritic cells (pDCs). Binding of deoxyribonucleic acid (DNA) from the NETs to TLR9 produces more IFNα, which, in turn, can result in excessive NET production. IFN pathways result in both a proinflammatory response and immunoregulation. Although neutrophils are integral for hosts to successfully eliminate viral infections, certain viruses have adapted mechanisms to hijack the IFN response to cause unwanted neutrophil-induced host damage. Excessive production of cytokines can lead to fatal immune-mediated overreactions to the viral threat. Other abbreviations: CXCR2: CXC chemokine receptor 2, MMP: matrix metalloproteases, NAMPT: nicotinamide phosphoribosyltransferase, ROS: reactive oxygen species.
Figure 2. Interferon (IFN)-mediated regulation of neutrophils upon viral infection of a host. Viral infections induce many changes to neutrophil biology. Toll-like receptor (TLR) expression increases after viral exposure, resulting in increased production of type I interferons (IFNs). Neutrophils possess receptors for all three interferon subsets. A positive feedback loop occurs during the production of neutrophil extracellular traps (NETs). IFNα results in NET production, which in turn activates plasmacytoid dendritic cells (pDCs). Binding of deoxyribonucleic acid (DNA) from the NETs to TLR9 produces more IFNα, which, in turn, can result in excessive NET production. IFN pathways result in both a proinflammatory response and immunoregulation. Although neutrophils are integral for hosts to successfully eliminate viral infections, certain viruses have adapted mechanisms to hijack the IFN response to cause unwanted neutrophil-induced host damage. Excessive production of cytokines can lead to fatal immune-mediated overreactions to the viral threat. Other abbreviations: CXCR2: CXC chemokine receptor 2, MMP: matrix metalloproteases, NAMPT: nicotinamide phosphoribosyltransferase, ROS: reactive oxygen species.Certain viruses are capable of infecting neutrophils. During IAV infection, neutrophils initiate a multifaceted immune response. Type I IFNs are expressed, along with ISGs and upregulation of PRRs [13]. Viral entry is required for this to occur, but replication is not essential. The virulent H3N2 influenza strain also infects neutrophils and induces a robust type I IFN signaling and regulatory response starting at three hours post-infection [11]. Lungs experiencing viral infection have a different immunological environment compared to bacterial lung infections, composed of type I IFNs and their resulting ISGs. It thus follows that neutrophils entering this virus-conditioned microenvironment would respond differently than they would to a bacterial infection. Viruses also possess genes to suppress type I IFNs to mediate their survival. IAVs express a nonstructural protein (NS1) that prevents induction of IFN-β [86]. Experiments in ferrets using NS1 from the pandemic-causing strain of IAV from 1918 determined that this protein significantly delayed the type I IFN response [87]. Moreover, the USSR/90/77 strain of IAV mediated a less pronounced delay in the IFN response. Additional research in ferrets showed mild influenza infections had robust innate responses, while severe disease was associated with reduced type I and type II IFN responses [88]. A genetically altered variant of IAV with NS1 deleted restored the IFN-α and IFN-β responses, coupled with increased NF-κB activation [89]. Influenza virus-infected neutrophils initiated the adaptive immune system by transitioning into antigen-presenting cells and subsequently activating effector antiviral CD8+ T cells [90]. Neutrophil depletion decreased the magnitude of virus-specific CD8+ T cells, although it did not impact T cell trafficking in the context of a pulmonary influenza infection [91].
Type I IFNs are integrally intertwined in most aspects of neutrophils’ existence, mediating both neutrophil production and cellular regulation. Type I interferons regulate nicotinamide phosphoribosyltransferase (NAMPT) signaling, which, in turn, is involved in survival and maturation of neutrophils [92]. Specifically, IFNs suppress NAMPT, as demonstrated in IFN-deficient animal models. Deficiency of IFN leads to an increase in NAMPT during neutrophil progenitor maturation in the bone marrow. During development, NAMPT increases early progenitor survival and later slows down neutrophil differentiation. During later life stages, when mature neutrophils are recruited to infected regions, IFN—alongside G-CSF and TNF—prolongs neutrophil survival [4]. Interferon-α delays neutrophil apoptosis by inducing cellular inhibitors of apoptosis 2 (cIAP2) via STAT in a similar manner to G-CSF [93]. Synthesis of cIAP2 is dependent on Janus kinase 2-STAT3 activation. Type I IFNs downregulate G-CSF, which is involved throughout the neutrophil lifecycle [94,95,96]. G-CSF causes STAT3-dependent changes within the bone marrow, influencing neutrophil migration [97]. By downregulating key neutrophil migratory control signals, IFN production can control the magnitude of neutrophil-mediated responses to viral infections. Interferon-β initiates phosphatidylinositol-3 kinase-dependent survival for neutrophils, thus preventing apoptosis [98]. In the context of cancers, IFN-β is needed to maximize neutrophil cytotoxicity [95].
Neutrophils can produce type II IFNs under multiple conditions. During renal ischemia-reperfusion injuries, neutrophils produce IFN-γ, a phenomenon that is dependent upon activation of natural killer-T cells in the kidneys within three hours of reperfusion [99]. IFN-γ is also produced by Gr-1+CD11b+ cells in the context of early islet graft rejection of the pancreas, which are again reliant on NKT cells [100]. Pathogens are also capable of initiating IFN-γ responses by neutrophils, as observed in Streptococcus pneumoniae experiments [101]. Clearance of pathogens, mediated by neutrophil-derived IFN-γ, is reliant on nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, Ras-related C3 botulinum toxin substrate 2 (Rac2), and Hck/Lyn/Fgr Src family tyrosine kinases. Type II IFN production within neutrophils requires these compounds to be produced. NETs are a proposed clearance mechanism [102]. Detailed analyses have illustrated that MyD88 is also critical for IFN-γ production by neutrophils, although TLRs and TRIF are not apparently involved [101]. Neutrophils subsequently can respond to IFN-γ by upregulating expression of genes and oxidative burst capabilities. Clearly, the traditional definition of neutrophils as terminal phagocytes has been altered by research demonstrating fine-tuned neutrophil protein synthesis in response to external stimuli [103].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094726
This entry is offline, you can click here to edit this entry!