The emergency caused by SARS-CoV-2 had, and still has, devastating socio-economic aspects. Assessing the impact of COVID-19 on vulnerable groups of people is crucial for the adaptation of governments’ responses. Growing scientific evidence suggests that it is essential to keep the attention on people after acute SARS-CoV-2 infection; indeed, some clinical manifestations are frequently present even after recovery. There is consensus on the need to define which symptoms persist after the infection and which disabilities may arise after COVID-19. Recent reviews, case reports, and original contributions suggest that various organs may be affected, and neurological symptoms are present in about one third of patients with COVID-19. Neurological complications after severe COVID-19 infection might include delirium, brain inflammation, stroke, and nerve damage. In the recent pandemic, neurologists and neurobiologists have a chance to study key features of infection neurology.
- COVID-19
- SARS-CoV-2
- neurology
- brain damage
- post-scute COVID-19 neurological syndrome
1. Neurobiology of SARS-CoV-2 Infection
Several studies recently showed that symptoms affecting the nervous system, such as sensory disturbances, headache, nausea, and vomiting, may occur in patients with SARS-CoV-2 infection and persist after infection [1]. Here, we discuss the different neurobiological processes and mechanisms linking SARS-CoV-2 to neuronal dysfunction.
2. Potential Mechanisms for the Penetration of SARS-Cov2 in CNS
The potential pathways of SARS-CoV-2 neuro-invasion from the periphery to the brain are many. All these routes are determined by the angiotensin-converting enzyme-2 receptor (ACE2), used by SARS-CoV-2 to bind and penetrate human cells [2]. This receptor is broadly distributed in lungs, heart, liver, kidney, intestine, and oral and nasal mucosa.
2.1. Direct Route
The existence of the direct route is hypothesized with the aid of coronavirus-infected animal experiments [3]. Possibly, after the droplets containing SARS-CoV-2 reach the nasal cavity, most viruses head to the lung, while others adhere to the mucosa of the nasal cavity from which they directly attack the olfactory sensory neurons in the olfactory epithelium; from here, they can be transferred into the CNS through the olfactory nerve. In addition, the abundant capillary blood vessels and lymphatics existent in the nasal mucosa provide chances for virus invasion of the bloodstream, from which they reach the brain. Moreover, SARS-CoV-2, adhering to the nasal mucosa or accessing the eye conjunctiva, may also reach the trigeminal nerve and then the brain through the route used for brain drug delivery [4]. The virus may also infect the sensory neurons in the taste buds, reach the nucleus of the solitary tract (VII, IX, and X) or the trigeminal nerve (V), and enter the CNS through neuronal retrograde transport [4].
Recently, Song and collaborators suggested the direct neuro-invasion of SARS-CoV-2 in the central nervous system in human and mouse brains. They showed that the brain is a site for high SARS-CoV-2 replication, with metabolic changes in neurons (neuronal death) in human brain organoids. Neuronal infection can be excluded by blocking ACE2, with antibodies, or by administering cerebrospinal fluid from a COVID-19 patient. Furthermore, using mice overexpressing human ACE2, the same authors demonstrated SARS-CoV-2 neuro-invasion in vivo. Finally, using COVID-19 brain autopsy of deceased patients, the authors detected SARS-CoV-2 in cortical neurons [5].
2.2. Indirect Route via Bloodstream
Viruses, once entered in the lungs, flow into the bloodstream through ACE2 receptor, present in the epithelium of the respiratory system. Moreover, they can also enter the gastrointestinal tract and then reach the brain through the vasculature system [2]. Once it has entered the bloodstream, the virus can infect the endothelial cells of the blood–brain barrier (BBB) or the blood–cerebrospinal fluid barrier (BCSFB), and then disseminate toward the CNS via ACE2 receptors. Besides, SARS-CoV-2′s cytokine storm in severely affected patients damages the BBB, resulting in infiltration of inflammatory factors and other blood contents, including viral particles, in the CNS [5]. Viruses in the blood can also directly enter the fourth ventricle through a damaged BCSFB.
3. SARS-CoV-2: Cellular Mechanism
Spike (S) proteins are essential for the virus to enter into the host cells. These proteins bind to the cellular ACE2 receptor, which is also present in neurons [6]. Protein S is the surface glycoprotein of the virus responsible for its crown shape. This protein is composed by two subunits, S1 and S2 [3]. The S1 subunit consists of the N-terminal domain (NTD) and the C-terminal domain (CTD). The receptor-binding domain (RBD) in the CTD is responsible for binding to the host cell. The S2 subunit allows the fusion with membranes. Full-length protein S, RBD domain, S1 subunit, and NTD are used as antigens to develop SARS-CoV-2 vaccines, including adenoviral, RNA-based, DNA-based, and protein subunit vaccines [7].
Furthermore, after intravenous injection, radioiodinated S1 (I-S1) protein was shown to cross the BBB in mice, be absorbed by brain regions, and enter the parenchymal brain space. Intranasally administered I-S1 penetrated the brain, but the levels were approximately ten times lower than those observed after intravenous administration. I-S1 intersected the BBB by transcytosis, and ACE2 was involved in the brain uptake [8].
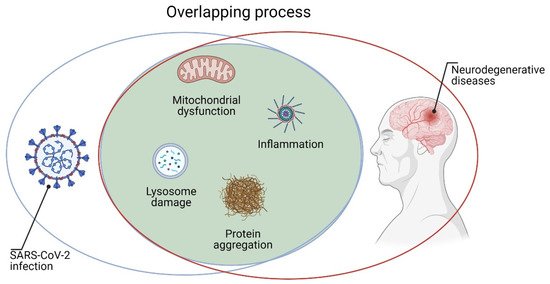
The penetration of SARS-CoV-2 into neurons can alter their cellular processes for energy production and protein folding [7]. Inside the cell, the virus can cause mitochondrial dysfunction and lysosome damage by inducing increased reactive oxygen species (ROS), protein misfolding/aggregation, and, ultimately, cell death [5]. It is important to underline that all these processes are involved in the pathogenesis of various neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [9][10][11]. Moreover, binding of ACE2 to the SARS-CoV-2 spike protein can reduce the conversion of angiotensin 2 (AT2) to AT [12]. Higher levels of AT2 are associated with pro-inflammatory markers and vascular injury to brain cells and other organs, all processes involved in neurodegeneration [12]. Furthermore, high levels of inflammation (cytokine storm) and BBB lesions in the brain are very likely to have long-term consequences on neurodegeneration. Indeed, there is indication that brain inflammation may contribute to the pathology of neurodegenerative diseases, including AD, PD, and ALS [13]. Coronaviruses such as SARS-CoV-2 could remain inside neurons without inducing toxic effects [14]. Therefore, in patients with acute SARS-CoV-2 infection, the virus could theoretically cause brain degeneration decades later [9]. Consequently, it would be useful to follow patients who were affected by COVID-19, allowing one to establish the real relationship between viral infection and neurodegenerative disorders (Figure 1). Finally, a better understanding of cellular and molecular mechanisms through which CoVs induced neuronal damage could help in performing new therapeutic strategies.
4. Neuroimmunology of COVID-19 Infection
Viruses stimulate the innate immune cells that recognize the molecular patterns associated with the pathogens. Decrease of the efficiency of the innate immune responses in eliminating the virus leads to the activation of adaptive immune system. The immune cells secrete cytokines and/or chemokines such as interleukin-6 (IL-6), interferon-γ (IFN-γ), interferon-γ-inducible protein-10 (IP-10), and monocyte chemoattractant protein-1 (MCP)-1. These molecules promote the influx of monocytes/macrophages and neutrophils to the site of infection [15]. Generally, this response is able to eliminate the virus, but occasionally the immune system is dysregulated, which leads to the alteration of the immune homeostasis [16]. SARS-CoV-2 infection can cause an intense inflammatory response and cytokine storm, which promotes lung pathogenesis and respiratory failure. This condition is one of the reasons for the higher mortality rate in fragile populations. As previously described, during COVID-19, the immune system is anomalously involved, and this may exacerbate autoimmunity in genetically predisposed people. Abnormal activation of immunological pathways could trigger mechanisms that alter the molecular recognition between viral epitopes and autoepitopes. Exacerbated immune-mediated manifestations are described in COVID-19 patients and represent the first sign of infection in fragile individuals. In addition, a robust adaptive immune response is essential for a conclusive viral clear and avoiding a re-infection [16].
During severe infection, the SARS-CoV-2 is able to infect macrophages, microglia, and astrocytes in the brain, which are particularly important. This neurotropic virus can stimulate glial cells and induce a pro-inflammatory status [17]. The most important cytokine during COVID-19 is interleukin 6 (IL-6), which is positively correlated with the severity of COVID-2019 symptoms. High levels of proinflammatory cytokines can cause confusion and alteration of consciousness [18]. A high level of inflammation may be associated with thrombophilia and increase the risk of stroke and other thrombotic events. The complementary activation may additionally lead to thrombotic microvascular injury in patients with severe COVID-19 [19]. Mohamud and coworkers reported five events of acute ischemic stroke simultaneous or 14 day delayed in patients with SARS-CoV-2 infection [20]. The mechanism of this event can be explained considering that the virus induces a high infiltration of macrophages, neutrophils, and T cells in the atheromatous plaques present in the blood vessel, exposing the patient to a thrombotic event. This can occur at the level of the carotid arteries [21]. The inflammation caused by SARS-CoV-2 infection can cause thinning of their fibrous caps, invasion of lipids, and expansion of the lipid layer [20].
High levels of cytokine and chemokine liberation may additionally lead to brain injury through microglial activation. In fact, in a case report, marked microglial nodules and neuronophagia were found in the brain tissue of a patient with COVID-19 [22].
5. Clinic Manifestation of Neuropathology of COVID-19 Infection
Neurological manifestations of COVID-19 infection are increasing to date. Ellul and collaborators elucidated the clinical features associated with SARS-CoV-2 infection in either central or peripheral nervous systems. Usually, neuropathological manifestations appear at the same time as the viral respiratory infection or some days later. Unfortunately, early reports did not have sufficient details to detect the presence of obvious neurological manifestations [23]. As the pandemic progresses, reports of neurological manifestations are increasing. To date, a multicenter study reported neurological manifestation as follows: three patients with encephalitis, 69 patients with other encephalopathies, and three patients with encephalomyelitis. Information relative to these patients is limited, but three died and, in three patients, neurological symptoms persisted long enough to require rehabilitation. Regarding peripheral nervous system manifestations, the study reports: seven patients with Guillain-Barrè disease diagnosed at the beginning of the viral infection and five patients with other neuropathies including Rhabdomiolysis. Among these patients, three needed rehabilitation, another three reported a longer time in intensive unit, and the rest reported minor or no information [24]. Furthermore, in 34 patients with cerebrovascular disease (ischaemic stroke and attacks), 20 died and six needed rehabilitation [23]. Loss of smell (anosmia) and taste (ageusia) emerged as common symptoms of COVID-19 and were present in 357 patients out of 417 in a European study [25] and in 68 out of 259 patients in a French retrospective observation [26]. There was no information on patients with abnormal smell and taste in persistent neurological symptoms. In the study of Varatharaj and colleagues at the beginning of 2020, among 125 patients recruited, the following percentage reported neurological problems: 13% with encephalopathy, 18% with a neuropsychiatric diagnosis (psychosis, neurocognitive syndrome, and an affective disorder), and 62% patients with cerebrovascular events [27].
In a pediatric multicenter neurological study, Lindan and collaborators found that the entire cohort studied did well with COVID-19 infection, whereas all children with hospital-acquired co-infections died, and two children were severely impaired and needed rehabilitation [28].
Although, the percentage of patients needing rehabilitation due to neurological associated COVID-19 manifestation is low, this is certainly due to the lack of follow up description after the hospital period of infection. At the beginning of the pandemic period, several observational studies lacked neurological description, implying that neurological manifestations were not evident. Today, it is quite clear that neurological traits associated with COVID-19 infection can be considered a sequel of the viral infection. Rogers and collaborators in a systematic review and meta-analysis showed how anxiety, fatigue, depression and post-traumatic stress were present in people surviving the COVID-19 infection as long-term manifestation. The etiology of the outcomes of infection with COVID-19 is likely to be multifactorial and might include the direct effects of viral infection (including encephalitis), cerebrovascular disease, physiological impairment (such as hypoxia), immunological activation, social isolation, and psychological impact. Many survivors complain about memory, attention, concentration, or mental impairments even after one year [29]. Eventually, it is quite possible that the psychiatric outcomes could be unrelated to the COVID-19 infection and may rather be a consequence of literature selection bias. At the same time, literature reporting post-illness studies did not agree on follow-up times and the studies cannot be compared. Therefore, thus far, there are too few studies for drawing conclusions but there is enough information to hypothesize that, in select populations, COVID-19 infection might drive neurological and psychiatric drawbacks.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10091947
References
- Nuzzo, D.; Picone, P. Potential neurological effects of severe COVID-19 infection. Neurosci. Res. 2020, 158, 1–5.
- Li, Z.; Liu, T.; Yang, N.; Han, D.; Mi, X.; Li, Y.; Liu, K.; Vuylsteke, A.; Xiang, H.; Guo, X. Neurological manifestations of patients with COVID-19: Potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front. Med. 2020, 14, 533–541.
- Li, K.; Wohlford-Lenane, C.; Perlman, S.; Zhao, J.; Jewell, A.K.; Reznikov, L.R.; Gibson-Corley, K.N.; Meyerholz, D.K.; McCray, P.B., Jr. Middle east respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J. Infect. Dis. 2016, 213, 712–722.
- Lochhead, J.J.; Kellohen, K.L.; Ronaldson, P.T.; Davis, T.P. Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci. Rep. 2019, 9, 1–9.
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.-E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135.
- Magrone, T.; Magrone, M.; Jirillo, E. Focus on receptors for coronaviruses with special reference to angiotensin-converting enzyme 2 as a potential drug target—A perspective. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 807–811.
- Karpiński, T.M.; Ożarowski, M.; Seremak-Mrozikiewicz, A.; Wolski, H.; Wlodkowic, D. The 2020 race towards SARS-CoV-2 specific vaccines. Theranostics 2021, 11, 1690–1702.
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat. Neurosci. 2021, 24, 368–378.
- Lippi, A.; Domingues, R.; Setz, C.; Outeiro, T.F.; Krisko, A. SARS-CoV -2: At the crossroad between aging and neurodegeneration. Mov. Disord. 2020, 35, 716–720.
- Di Carlo, M.; Giacomazza, D.; Picone, P.; Nuzzo, D.; San Biagio, P.L. Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Radic. Res. 2012, 46, 1327–1338.
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell. Longev. 2014, 2014, 1–11.
- Fotuhi, M.; Mian, A.; Meysami, S.; Raji, C.A. Neurobiology of COVID-19. J. Alzheimer’s Dis. 2020, 76, 3–19.
- Nuzzo, D.; Picone, P.; Caruana, L.; Vasto, S.; Barera, A.; Caruso, C.; Di Carlo, M. Inflammatory mediators as biomarkers in brain disorders. Inflammation 2013, 37, 639–648.
- Nath, A. Neurologic complications of coronavirus infections. Neurology 2020, 94, 809–810.
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362.
- Ahmadpoor, P.; Rostaing, L. Why the immune system fails to mount an adaptive immune response to a COVID-19 infection. Transpl. Int. 2020, 33, 824–825.
- Li, Y.; Fu, L.; Gonzales, D.M.; Lavi, E. Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J. Virol. 2004, 78, 3398–3406.
- Koralnik, I.J.; Tyler, K.L. COVID -19: A Global Threat to the Nervous System. Ann. Neurol. 2020, 88, 1–11.
- Connors, J.M.; Levy, J.H. Thromboinflammation and the hypercoagulability of COVID-19. J. Thromb. Haemost. 2020, 18, 1559–1561.
- Mohamud, A.Y.; Griffith, B.; Rehman, M.; Miller, D.; Chelb, A.; Chebl, A.; Patel, S.C.; Howell, B.; Kole, M.; Marin, H. Intraluminal carotid artery thrombus in COVID-19: Another danger of cytokine storm? Am. J. Neuroradiol. 2020, 41, 1677–1682.
- Wijeratne, T.; Gillard-Crewther, S.; Sales, C.; Karimi, L. COVID-19 pathophysiology predicts that ischemic stroke occurrence is an expectation, not an exception—A systematic review. Frontiers in Neurology. Front. Neurol. 2021, 11, 607221.
- Al-Dalahmah, O.; Thakur, K.T.; Nordvig, A.S.; Prust, M.L.; Roth, W.; Lignelli, A.; Uhlemann, A.-C.; Miller, E.H.; Kunnath-Velayudhan, S.; del Portillo, A.; et al. Neuronophagia and microglial nodules in a SARS-CoV-2 patient with cerebellar hemorrhage. Acta. Neuropathol. Commun. 2020, 8, 147.
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783.
- Sadeghi, A.; Tahmasebi, S.; Mahmood, A.; Kuznetsova, M.; Valizadeh, H.; Taghizadieh, A.; Nazemiyeh, M.; Aghebati-Maleki, L.; Jadidi-Niaragh, F.; Abbaspour-Aghdam, S.; et al. Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J. Cell. Physiol. 2021, 236, 2829–2839.
- Lechien, J.R.; Chiesa-Estomba, C.M.; De Siati, D.R.; Horoi, M.; Le Bon, S.D.; Rodriguez, A.; Dequanter, D.; Blecic, S.; El Afia, F.; Distinguin, L.; et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study. Eur. Arch. Oto Rhino Laryngol. 2020, 277, 2251–2261.
- Bénézit, F.; Le Turnier, P.; Declerck, C.; Paillé, C.; Revest, M.; Dubée, V.; Tattevin, P.; Arvieux, C.; Baldeyrou, M.; Chapplain, J.-M.; et al. Utility of hyposmia and hypogeusia for the diagnosis of COVID-19. Lancet Infect. Dis. 2020, 20, 1014–1015.
- Varatharaj, A.; Thomas, N.; A Ellul, M.; Davies, N.W.S.; A Pollak, T.; Tenorio, E.L.; Sultan, M.; Easton, A.; Breen, G.; Zandi, M.; et al. UK-wide surveillance of neurological and neuropsychiatric complications of COVID-19: The first 153 patients. Lancet Psychiatry 2020, 7, 875–882.
- Lindan, C.E.; Mankad, K.; Ram, D.; Kociolek, L.K.; Silvera, V.M.; Boddaert, N.; Stivaros, S.M.; Palasis, S. Neuroimaging manifestations in children with SARS-CoV-2 infection: A multinational, multicentre collaborative study. Lancet Child Adolesc. Health 2021, 5, 167–177.
- Rogers, J.P.; Chesney, E.; Oliver, D.; A Pollak, T.; McGuire, P.; Fusar-Poli, P.; Zandi, M.S.; Lewis, G.; David, A.S. Psychiatric and neuropsychiatric presentations associated with severe coronavirus infections: A systematic review and meta-analysis with comparison to the COVID-19 pandemic. Lancet Psychiatry 2020, 7, 611–627.