The identification of thrombospondin-1 as an angiogenesis inhibitor in 1990 prompted interest in its role in cancer biology and potential as a therapeutic target. Decreased thrombospondin-1 mRNA and protein expression are associated with progression in several cancers, while expression by nonmalignant cells in the tumor microenvironment and circulating levels in cancer patients can be elevated. THBS1 is not a tumor suppressor gene, but the regulation of its expression in malignant cells by oncogenes and tumor suppressor genes mediates some of their effects on carcinogenesis, tumor progression, and metastasis. In addition to regulating angiogenesis and perfusion of the tumor vasculature, thrombospondin-1 limits antitumor immunity by CD47-dependent regulation of innate and adaptive immune cells. Conversely, thrombospondin-1 is a component of particles released by immune cells that mediate tumor cell killing. Thrombospondin-1 differentially regulates the sensitivity of malignant and nonmalignant cells to genotoxic stress caused by radiotherapy and chemotherapy.
1. Introduction
Cancer has been described as a wound that does not heal [
1]. Despite many similarities between the wound and tumor microenvironments, exploitable differences in the recruited cell types and their secreted extracellular matrix products have been identified as contributors to cancer progression and potential therapeutic targets. Our interest in studying the role of the secreted protein thrombospondin-1 (TSP1, encoded by
THBS1) in the tumor microenvironment arose from our observation that TSP1 expression decreased during malignant progression in melanoma and breast carcinoma cell lines. The expression of oncogenic forms of K-Ras, H-Ras, and N-Ras in bronchial epithelial cells also suppressed TSP1 mRNA and protein expression [
2]. Conversely, re-expression of TSP1 impaired tumor growth and metastasis in several types of cancer, including melanoma, glioblastoma, prostate carcinoma, squamous cell carcinoma, and cervical carcinoma [
3,
4,
5,
6,
7]. Other oncogenes were subsequently found to negatively regulate TSP1 expression, including
MYC [
8,
9]. Conversely, inactivation of tumor suppressor genes such as
TP53 and
RB1 suppressed TSP1 expression [
10,
11,
12,
13,
14,
15]. Deletion of
THBS1 is a rare event in most human cancers, and the observed loss of expression largely results from epigenetic effects of the altered oncogenes and tumor suppressor genes [
16]. Despite the general loss of TSP1 expression in malignant cells, elevated circulating levels of TSP1 in blood have been reported in several human and murine cancers [
10]. TSP1 expression is also induced in the wound microenvironment [
17]. The relevance of TSP1 in the wound/cancer dichotomy was further suggested by a report that showed TSP1 mRNA is upregulated in renal tissue regeneration but downregulated in renal cell carcinoma [
18].
In addition to an intrinsic role for TSP1 expressed by tumor cells, increased growth of B16 melanomas and F9 testicular teratocarcinomas was observed when implanted in syngeneic mouse strains lacking
Thbs1 [
19]. As TSP1 is a secreted protein, its abundance in the tumor microenvironment depends on both tumor and stromal cell expression. TSP1 in the tumor microenvironment can influence the behavior of multiple cell types that regulate tumor growth and metastasis. In addition to being regulated by oncogenes and tumor suppressor genes, TSP1 in the microenvironment can mediate feedback regulation of their expression, as demonstrated for p53 and Myc [
20]
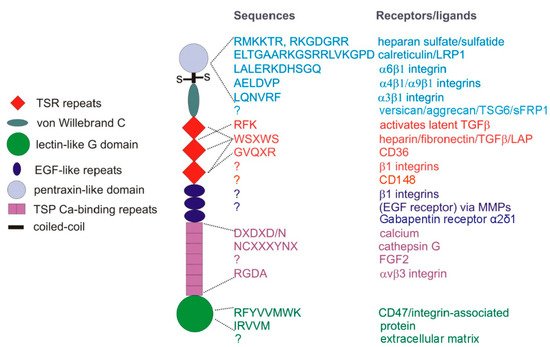
Consistent with the complexity of function for other matricellular proteins, both protective and tumor-promoting functions of TSP1 have also been reported. Divergent roles of TSP1 can be mediated by engaging different TSP1 receptors (). In cases where cells express multiple TSP1 receptors, responses to TSP1 can be biphasic. For example, by engaging several integrins, TSP1 can promote endothelial cell motility and proliferative responses, whereas engaging CD47 on the same cells inhibits the same responses [
10,
21,
22].
Figure 1. TSP1 subunit domains and their cell surface receptors or extracellular ligands. TSP1 is a ~450 kDa homotrimer of subunits linked by disulfide bonds near the N-terminal pentraxin-like domain. Type 1 TSP1 repeats (TSR), EGF-like, and calcium-binding repeats form the central stalk region of TSP1, connecting the N- and C-terminal globular domains.
2. TSP1 Regulation of Angiogenesis and Tumor Perfusion
2.1. Inhibition and Stimulation of Angiogenesis
Early efforts to define the underlying mechanism by which the loss of TSP1 in the tumor microenvironment contributes to cancer progression resulted in three independent reports in 1990 identifying TSP1 as an angiogenesis inhibitor [
21,
73,
74]. TSP1 is a potent inhibitor of endothelial cell migration and proliferation and an inducer of endothelial apoptosis [
21,
75]. Consistent with these results, TSP1 inhibited angiogenesis in rat cornea [
74], chicken chorioallantoic membrane [
76], and muscle explants in 3D culture [
77]. Evidence from mouse models demonstrated that elevating TSP1 expression inhibits tumor growth with a corresponding decrease in vascular density [
3,
11,
78].
CD36 was the first TSP1 receptor identified to mediate its anti-angiogenic activity [
35], and peptide mimetic drugs derived from a sequence in the second TSR repeat of TSP1 that binds to CD36 () have been developed to inhibit tumor angiogenesis [
43]. However, some observations were not consistent with the hypothesis that TSP1 functions solely as an angiogenesis inhibitor. If true, one would expect TSP1 to be downregulated during wound repair as well as in cancer. One would also predict that the loss of this inhibitor would accelerate wound repair in a
Thbs1−/− mouse. Instead, TSP1 was rapidly up-regulated following excisional skin wounding [
79], and the
Thbs1−/− mouse exhibited delayed wound closure in this model [
80]. Furthermore, TSP1 antisense oligonucleotides delayed repair of excisional skin wounds in wild-type (WT) mice [
79]. This activity of TSP1 contrasts with TSP2, a closely related protein that also inhibits angiogenesis and engages CD36 [
81,
82,
83]. TSP2 is expressed only late in wound closure, and
Thbs2−/− mice showed the predicted accelerated excisional wound repair [
80]. Agah et al. rationalized the unexpected wound repair phenotype of the
Thbs1−/− mouse by invoking another TSP1 responsive cell type. They found that macrophage infiltration was impaired in the absence of TSP1, and chemokines’ levels essential for wound repair such as MCP1 were lower. Therefore, they proposed that the dominant activity of TSP1 induced early in excisional wound repair is not to limit angiogenesis but instead to recruit monocytes into the wound.
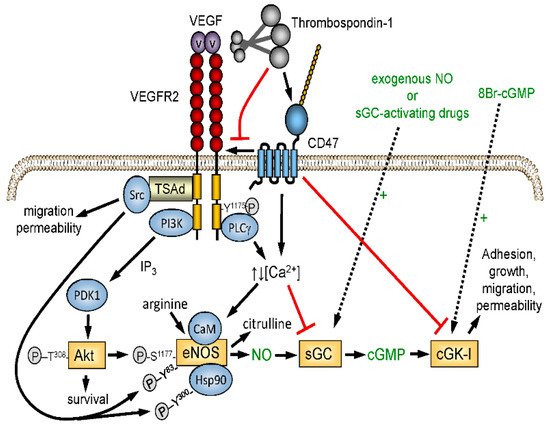
The excisional skin wound model lacked ischemic stress, which reveals a more critical function of TSP1 in limiting the angiogenic response required for wound repair. Nitric oxide (NO)/cGMP signaling is a central regulator of angiogenesis and tissue perfusion under ischemia, and TSP1 signaling through its receptor CD47 potently inhibits NO biosynthesis and signaling in vascular cells [
37] ().
Thbs1−/− and
cd47−/− mice exhibit enhanced vascular responses to NO and improved recovery from ischemic injuries [
84]. Blocking CD47 expression or function in WT mice, rats, and miniature pigs improved recovery from ischemic injuries [
85,
86,
87,
88,
89,
90]. TSP1 inhibition of the angiogenic response to NO donors was lost in muscle explants from
cd47−/− mice but preserved in
cd36−/− mice [
91]. We established physiological and pathophysiological roles for CD47 in mediating TSP1 signaling in endothelial cells, vascular smooth muscle cells, and platelets [
77,
84,
92,
93]. Therefore, the elevated TSP1 in wounds has two acute activities in addition to its long-term regulation of angiogenesis and vascular remodeling: (1) TSP1 is a potent vasoconstrictor that acutely limits bleeding but can be counterproductive for tissue survival under ischemic stress, and (2) TSP1 is an autocrine factor released by platelets that promotes hemostasis.
Figure 2. Redundant regulation of NO signaling by TSP1–CD47 interaction. Endogenous NO synthesis is stimulated via Akt-mediated phosphorylation of endothelial nitric oxide synthase (eNOS) downstream of the VEGF receptor VEGFR2. Ligation of CD47 by TSP1 also inhibits activation of soluble guanylyl cyclase (sGC) mediated by endogenous or exogenous NO. TSP1 also inhibits calcium-dependent activation of eNOS and NO signaling downstream of cGMP by inhibiting cGMP-dependent protein kinase (cGK-1).
These studies provide additional insights into why TSP1 is downregulated in cancers. As TSP1 limits angiogenesis and perfusion, its local expression is clearly a disadvantage to a growing tumor [
10]. Loss of TSP1 expression may also decrease the thrombogenic potential of the tumor vasculature by maximizing the anti-thrombotic activity of NO produced by the tumor and its stroma.
2.2. Vascular Perfusion of Tumors and the Steal Effect
CD47-dependent antagonism of NO signaling by TSP1 limits the perfusion of healthy tissues, but this signaling is impaired in the tumor vasculature [
94]. In the closed system of vascular physiology, vasodilation in one vascular bed can “steal” blood flow from another vascular bed fed by the same perfusing artery [
95]. Conversely, TSP1-mediated constriction of healthy tissue vasculature can result in increased perfusion of tumors, providing selective pressure for the elevated circulating TSP1 levels observed in some cancers [
10].
2.3. Endothelial Cell Apoptosis
TSP1 and its type 1 repeats induce apoptosis of endothelial cells [
75]. CD36 and CD47 have been implicated in mediating TSP1 apoptotic signaling [
96,
97]. The induction of TNFα expression by TSP1 in endothelial cells may mediate cell death based on a requirement for tumor necrosis factor receptor-1 [
98]. In the tumor microenvironment, TSP1-induced endothelial cell apoptosis contributes to the antitumor activity of low-dose cyclophosphamide treatment [
99].
3 TSP1 and Antitumor Immunity
3.1. Regulation of T Cell Immunity
TSP1 globally suppresses changes in gene expression that are induced by T cell antigen receptor (TCR) signaling [
100], which requires CD47 [
25,
51,
101]. Engaging CD47 limits T cell-dependent inflammation in vivo [
102], induces T cell apoptosis in the context of TCR signaling [
103], and induces CD4
+ T cell differentiation into regulatory T cells [
104]. The latter study implied that TSP1 has the same activity, but the only evidence presented involved a TSP1 peptide analog with limited specificity [
37,
105].
Immune cell responses to TSP1 are defined by integrating signals from several cell surface receptors [
25,
106,
107,
108]. Jurkat T cells have been a valuable model for defining such cross-talk because somatic mutants are available that lack the TSP1 receptors α4β1 integrin, CD47, and several signaling molecules downstream of these receptors [
25,
107]. These tools and T cells isolated from transgenic mice lacking TSP1 or CD47 enabled us to confirm or exclude the contribution of each receptor to specific T cell responses in vitro and immune responses in vivo [
25,
51,
101,
107,
109,
110].
TSP1-mediated CD47 signaling limits antigen-dependent T cell activation by several mechanisms. By inhibiting signal transduction downstream of the T cell receptor, TSP1 inhibits the expression of genes encoding IL-2 and TNFα that can stimulate proliferation and activation of other immune cells and the α-subunit of the IL-2 receptor (CD25), which limits the ability of T cells exposed to TSP1 to respond to exogenous IL-2. TSP1 also limits the expression of cystathionine β-synthase, which is induced during T cell activation and produces the diffusible mediator H
2S that acts on the T cell cytoskeleton to regulate T cell polarization required for immunological synapse formation [
101,
111].
Sensitivity of B16 melanomas in immune-competent mice to ablation by tumor irradiation was enhanced when either
Thbs1 or
Cd47 were disrupted in the tumor microenvironment [
112,
113]. Subsequent studies revealed that this increased sensitivity to ionizing radiation requires CD8
+ T cells. Furthermore, therapeutic blockade of TSP1/CD47 signaling using antibodies or antisense CD47 knockdown enhances antigen-dependent killing of irradiated tumor cells by mouse and human CD8
+ T cells in vitro and tumors in athymic mice following adoptive transfer of tumor-specific CD8
+ T cells [
113,
114]. Therefore, CD47 on CD8
+ T cells functions as an adaptive immune checkpoint that mediates TSP1-dependent inhibition of tumor cell killing.
3.2. TSP1 Regulation of Innate Immune Cells
TSP1 also regulates innate immune cells relevant to the tumor microenvironment. Early studies examined TSP1 effects on neutrophil oxidative burst and found that TSP1 synergizes with a formylated bacterial peptide to stimulate an oxidative burst response [
115,
116]. Others identified inhibitory TSP1 functions in NK cells [
117,
118], dendritic cells [
119,
120,
121,
122,
123], and monocytes [
108,
117,
124,
125,
126,
127]. Functions of TSP1 in other aspects of immune responses include modulation of TLR3-mediated inflammatory signaling in VSMC [
71].
Several studies have reported the regulation of myeloid cell functions by TSP1 in the tumor microenvironment. Secretion of TSP1 by tumor cells increased macrophage recruitment and increased M1 polarization in a melanoma xenograft model [
32]. TSP1 increased oxidative killing of tumor cells by macrophages in vitro. Tumors with low metastatic potential were reported to induce TSP1 expression by bone marrow-derived Gr1
+ myeloid cells, and targeted deletion of
Thbs1 in myeloid cells abolished their anti-metastatic activity [
128]. Myeloid-derived suppressor cells released EVs that contained TSP1, and a TSP1 antibody specifically inhibited migration of myeloid-derived suppressor cells induced by these EVs [
129]. Finally, a protective role of TSP1 to limit UVB-induced skin carcinogenesis was attributed to an anti-inflammatory function that limited the expansion of myeloid progenitor cells in the neutrophil lineage [
130].
Interest in CD47 as a regulator of anti-tumor innate immunity was heightened by reports that a CD47 blocking antibody enhances NK- and macrophage-mediated killing of tumor cells [
131,
132]. CD47 was originally discovered as a tumor-associated antigen for ovarian cancer [
133], but its pathophysiological significance was unclear. Elevated CD47 expression on tumor cells is now recognized as a general mechanism to evade host innate immunity for various solid tumors and hematologic malignancies [
131,
132,
134,
135,
136,
137,
138,
139,
140,
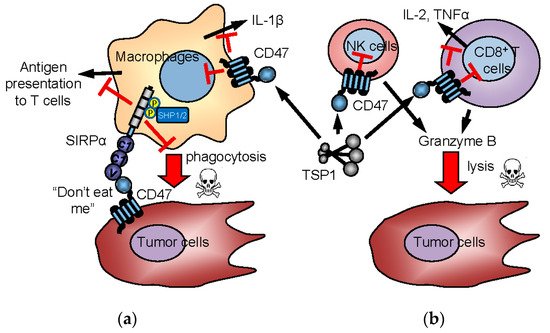
141]. The prevailing model proposes that high CD47 expression on tumor cells engages the inhibitory counter-receptor SIRPα on phagocytes to prevent tumor cell killing (a). CD47-induced SIRPα signaling in macrophages and dendritic cells indirectly enhances anti-tumor adaptive T cell immune responses [
142]. However, CD47 blockade can also enhance antibody-dependent cellular cytotoxicity independent of SIRPα signaling [
143]. Enhanced macrophage clearance of tumor cells damaged by irradiation was a plausible mechanism to account for the synergism we observed between radiation and CD47 knockdown in syngeneic tumor models [
144]. However, further studies revealed that CD47 signaling in murine and human CD8
+ T cells directly inhibits their antigen-dependent killing of tumor cells (b) [
113,
114].
Figure 3. Innate and adaptive immune checkpoint functions of CD47 and TSP1. In the “don’t eat me” model (a), CD47 on tumor cells interacts with SIRPα on macrophages to induce inhibitory signaling, preventing the phagocytic killing of tumor cells. CD47 is also expressed by immune cells and mediates TSP1 signaling in immune cells. TSP1 interaction with CD47 on NK cells and cytotoxic T cells inhibits their activation and limits granzyme B production that mediates antigen-dependent lysis of tumor cells (b). Inhibitory TSP1 signaling mediated by CD47 on macrophages and dendritic cells limits their presentation of antigens to T cells and limits macrophage production of IL-1β.
Conversely, in the absence of CD8
+ T cells, blocking CD47 does not inhibit tumor growth in syngeneic models [
113,
145,
146], and genetic disruption of SIRPα signaling in macrophages did not impair syngeneic tumor growth [
143]. Thus, complete loss of the “don’t eat me” signal is insufficient to prevent tumor growth in the absence of cytotoxic T cell activity or secondary stimuli to induce innate immune clearance [
147]. The tumor cell response to damage induced by ionizing radiation provides such a signal to enhance CD47-induced tumor clearance in immune-competent mice. This clearance also requires cytotoxic T cell activity [
113].
Several humanized CD47 antibodies and SIRPα decoys have entered clinical trials as cancer therapeutics [
54,
148,
149,
150,
151]. All clinical CD47 antibodies block SIRPα binding to CD47 and are intended to enhance innate immune-mediated clearance of tumors [
138,
140,
152,
153,
154]. Likewise, anti-SIRPα antibodies have shown preclinical activity as a single agent or in combination with a tumor-targeted agent [
54]. However, with respect to CD47-directed agents, several findings raise doubts about an exclusive role of SIRPα and macrophages in the antitumor activity of CD47 blockade. We and others have reported that CD47 antibodies exhibit anti-tumor activities independent of SIRPα signaling [
113,
143,
147,
155,
156,
157,
158].
3.3. CD47 and TSP1 Signaling in Macrophages
Recent studies have emphasized the role of the inhibitory receptor SIRPα in regulating macrophage phagocytosis of tumor cells that highly express CD47 [
141]. Still, macrophages also express CD47, and physiological concentrations of TSP1 limit the induction by lipopolysaccharide (LPS) of IL-1β mRNA and total IL-1β protein production by human macrophages [
159]. This inhibition could be explained by the ability of TSP1 binding to disrupt the interaction between CD47 and CD14, thereby limiting activation of NFκB/AP-1 by LPS. Only the CD47-binding domain of TSP1 exhibited this activity. In contrast, CD47, CD36, and integrin-binding domains of TSP1 independently enhanced the inflammasome-dependent maturation of IL-1β in human THP-1 monocyte-derived macrophages. Correspondingly, mouse bone marrow-derived macrophages lacking either TSP1 or CD47 exhibited diminished induction of mature IL-1β in response to LPS. Loss of CD47 also limited LPS induction of IL-1β, NLRP3, and caspase-1 mRNAs. These data demonstrate that TSP1 exerts CD47-dependent and -independent pro-and anti-inflammatory effects on the IL-1β pathway in macrophages.
3.4. Intrinsic Functions of CD47 in NK Cells
TSP1 and CD47 also have cell-intrinsic roles in regulating NK cells. TSP1 inhibited early NK cell proliferation and enhanced late expansion, but a role for CD47 was not examined [
117]. CD47 as a SIRPα counter-receptor enabled the engraftment of NK precursors in mice reconstituted with a human immune system [
160]. Treatment with an inhibitory CD47 antibody increased NK cell killing of human head-and-neck squamous carcinoma cells in vitro [
131]. However, NK cells were not known to express SIRPα, and the mechanism was unclear at the time. SIRPα expression was recently shown to be induced in activated NK cells, and ligation by CD47 alters NK cell function [
161]. Depletion of NK cells similarly attenuated the anti-tumor activity of a SIRPα blocking antibody in a syngeneic murine renal carcinoma model, but the same antibody did not inhibit NK killing of the tumor cells in vitro, further supporting a SIRPα-independent function of CD47 in NK cells [
162]. We found an increased abundance of lineage-negative cells within the spleen of
Cd47−/− mice and discovered these to be immature NK cells [
163]. This led us to further investigate the role of CD47 in NK cell homeostasis.
Cd47−/− mice exhibited depletion of NK precursors in bone marrow, consistent with the antiphagocytic function of CD47. In contrast, antisense CD47 knockdown or gene disruption resulted in a dose-dependent accumulation of immature and mature NK cells in spleen. Mature
cd47−/− NK cells exhibited increased expression of NK effector and interferon gene signatures and an increased proliferative response to interleukin-15 in vitro.
Cd47−/− mice were moderately impaired in controlling chronic Clone-13 LCMV infection. This was associated with depletion of splenic NK cells and loss of effector cytokine and interferon response gene expression in
cd47−/− NK cells. Broad CD47-dependent differences in NK activation, survival, and exhaustion pathways were observed in NK cell transcriptional signatures in LCMV infected mice. These data identify CD47 as a cell-intrinsic and systemic regulator of NK cell homeostasis and NK cell function in responding to a viral infection. Consistent with our data, a recent study found increased NK cell activation following induction of atherosclerosis in
cd47−/− mice [
118].
Extending these findings to the role of CD47 in cancer, we examined the NK cells in syngeneic B16 melanomas growing in WT versus
cd47−/− mice [
164]. Elevated CD47 expression in some cancers is associated with decreased survival and limited clearance by phagocytes expressing the CD47 counterreceptor SIRPα [
140,
141]. In contrast, we found that elevated CD47 mRNA expression in human melanomas is associated with improved survival [
164]. Gene-expression data identified a potential mechanism for this apparent protective function and suggested that high CD47 expression increases NK cell recruitment into the tumor microenvironment. The CD47 ligand TSP1 inhibited NK cell proliferation in vitro and the induction of CD69 expression [
164].
Cd47−/− NK cells correspondingly displayed augmented effector phenotypes, indicating an inhibitory function of CD47 on NK cells. Treating human NK cells with a CD47 antibody that blocks TSP1 binding abrogated its inhibitory effect on NK cell proliferation [
164]. Similarly, treating wild-type mice with a CD47 antibody that blocks TSP1 binding delayed B16 melanoma growth, associating with increased NK cell recruitment and increased granzyme B and interferon-γ levels in intratumoral NK but not CD8
+ T cells [
164].
However, B16 melanomas grew faster in
cd47−/− than in WT mice [
164]. Melanoma-bearing
cd47−/− mice exhibited decreased splenic NK cell numbers, with impaired effector protein expression and elevated exhaustion markers. Proapoptotic gene expression in
cd47−/− NK cells was associated with stress-mediated increases in mitochondrial proton leak, reactive oxygen species, and apoptosis [
164]. Global gene-expression profiling in NK cells from tumor-bearing mice identified CD47-dependent transcriptional responses that regulate systemic NK activation and exhaustion. Therefore, CD47 positively and negatively regulates NK cell function, and therapeutic antibodies that block inhibitory TSP1-CD47 signaling can enhance NK immune surveillance of melanomas.
3.5. TSP1 in Supramolecular Attack Particles
A novel role for TSP1 was recently identified in the supramolecular attack particles released by cytotoxic T cells and NK cells that kill target tumor cells [
165,
166]. A fragment of TSP1 was identified as a component of the outer shell of these particles that deliver perforin and granzyme-B from their core to kill target cells. Notably, CRISPR-mediated deletion of TSP1 impaired the cytotoxic activity of CD8
+ T cells, indicating that TSP1 plays an intrinsic role in the cytotoxic function of supramolecular attack particles.
4. TSP1 and Carcinogenesis
Several studies support roles for TSP1 in carcinogenesis. Targeted overexpression of TSP1 in the epidermis delayed and reduced premalignant epithelial hyperplasias induced by chemical carcinogens [240]. This protective role of TSP1 over-expression was extended to UVB-induced skin carcinogenesis [130] and spontaneous mammary adenocarcinomas in TgN-neu mice [241]. Conversely, loss of Thbs1 increased mammary adenocarcinomas in these mice [241] and increased osteosarcoma incidence but not the incidence of some other malignancies in mice lacking p53 [19]. In a murine model of prostate cancer metastasis to bone, increased TSP1 was observed in platelets, and implantation of tumors in Thbs1 null mice resulted in increased tumor size [242]. On the other hand, the absence of TSP1 reduced bone marrow-derived cell mobilization and enhanced osteoclast formation, resulting in decreased tumor-induced bone formation, suggesting a role of TSP1 in the pre-metastatic niche formation.
TSP1 limited angiogenesis and inflammatory responses that contribute to colorectal carcinogenesis in ApcMin/+ mice [243] and colorectal carcinogenesis induced by chronic inflammation [244]. The ability of TSP1 to regulate the responses of cells and tissues to stress prompted us to examine whether loss of TSP1 also has systemic effects on metabolism that modulate carcinogenesis [195]. ApcMin/+:Thbs1−/− mice exhibited decreased survival and higher tumor multiplicities in the small and large intestine relative to ApcMin/+ mice when fed a low-fat Western diet. However, the protective effect of endogenous TSP1 was lost when the mice were fed a high-fat Western diet. Biochemical profiles of liver tissue identified systemic metabolic changes associated with the effects of TSP1 and dietary lipid intake on tumorigenesis. A high-fat Western diet differentially regulated amino acid, energy, and lipid metabolism in ApcMin/+:Thbs1−/− mice relative to ApcMin/+ mice. Changes in ketone body and tricarboxylic acid cycle intermediates identified functional interactions between Apc and TSP1 signaling that control mitochondrial function. These data suggest that the protective role of TSP1 to limit adenoma formation in ApcMin/+ mice results in part from improved mitochondrial function and eicosanoid signaling [245].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094570