Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
As the most common primary liver cancer, HCC is a tricky cancer resistant to systemic therapies. The fibroblast growth factor family and its receptors are gaining more and more attention in various cancers. Noticing an explosion in the number of studies about aberrant FGF/FGFR signaling in HCC being studied, we were encouraged to summarize them.
- FGF
- FGFR
- HCC
1. Introduction
As one of the most common malignant tumors of the liver, hepatocellular carcinoma (HCC) is a major public health issue worldwide with increasing morbidity and cancer-related mortality but limited intervention options and low curative rates. Chronic liver disease associated with hepatitis B virus (HBV) or hepatitis C virus (HCV) is the most common etiology of HCC, especially in developing areas. Approximately 80% of HCC patients worldwide have HBV or HCV infection. Non-alcoholic fatty liver disease (NAFLD) and diabetes are the primary and increasing risk factors for HCC in developed countries. The consumption of aflatoxin B1, cigarettes, and alcoholic substances are also associated with HCC [1][2][3]. Although there are various kinds of interventions for HCC, only a few early-diagnosed patients can receive potential curative therapies through surgical resection, transarterial chemoembolization (TACE), or ablation. Even worse, HCC is a highly insidious cancer, meaning that HCC is often detected at intermediate or even advanced stages and therefore miss the optimal treatment window. In these cases, systemic pharmacological treatment is the best therapy but can only provide modest benefits [3][4]. Despite the many surveillance protocols, the overall survival of HCC is still unsatisfactory. Agent acquired resistance, the high recurrence rate after surgical resection, and the lack of biomarkers for HCC identification highlight the need for investigators to further elucidate the molecular pathology of HCC and provide more alternative therapeutic options.
The family of fibroblast growth factors (FGFs) consist of eighteen ligands and four homologous factors. FGFs bind with corresponding FGF receptors (FGFR1~4) and are involved in various actions, including early embryogenesis, angiogenesis, wound repair, metabolism, and many other physiological processes in adults [5][6]. The downstream effects of FGF/FGFR, such as regulating cell proliferation, differentiation, and survival, indicate that this axis is a potential target in the pathogenesis of multiple tumors. Several cancers, such as breast cancer, gastric cancer, endometrial cancer, bladder cancer, myeloma, and HCC, can be induced when FGF/FGFR signaling is abnormal [7][8][9][10][11]. Thus far, interventions targeting FGF/FGFR have provided modest benefits for patients, and some of these treatments have already been approved in the clinic. Furthermore, FGF/FGFRs show value as biomarkers for patient identification, which is essential for detecting the interpatient heterogeneity of HCC. All of the observations above indicate that targeting FGF/FGFR signaling is a promising therapeutic option for HCC patients.
2. The FGF/FGFR System
2.1. FGF, FGFR, and Co-Factor
FGF ligands are categorized into five paracrine subfamilies (FGF1, FGF2; FGF4, FGF5, FGF6; FGF3, FGF7, FGF10, FGF22; FGF8, FGF17, FGF18), one endocrine subfamily (FGF15/19, FGF21, FGF23) and several homologous factors (FGF11-14) (Table 1). FGF11-14 share substantial sequence homology with other FGFs, but they have neither recognizable secretory signal peptides nor the ability to be secreted from cells; thus, their bioactivity is intracellular and they cannot initiate any FGFR signaling pathways. Therefore, these molecules are not discussed in this review [12]. Mouse FGF15 is orthologous to human FGF19, sharing 51% amino acid identity [13].
Table 1. The classification of FGF ligands and their corresponding FGFRs.
FGFRs are divided into three domains as transmembrane receptors: three extracellular Ig-like loop domains (termed Ig I, Ig II, and Ig III), two intracellular tyrosine kinase domains (termed TK1-2), and a single transmembrane helix linking the extra and intra components. Ig-loop II and Ig-loop III are essential for signal transduction because they specifically recognize ligands and cofactors [14]. In contrast, a conserved stretch of 7–8 acidic residue sequences, called the acid box, is involved in receptor autoinhibition and signaling inhibition along with Ig I. Four FGFRs share high sequence similarities, especially FGFR1-3. Alternative splicing of FGFR is often observed in Ig-loop III of FGFR1-3, generating two isoforms generally named IIIb and IIIc (Figure 1). The FGFR IIIb isoform tends to be expressed in epithelial cells. While the FGFR IIIc isoform is more likely to be expressed on mesenchymal cells, the transformation from IIIb to IIIc is associated with epithelial-mesenchymal transition (EMT) [15]. FGFR4 lacks an alternative splicing exon and has no isoform. Given the core role of ligand recognition in Ig-loop III, the isoforms alter the ligand-receptor binding spectrum [15].
| FGF Subfamily | FGF | FGFR |
|---|---|---|
| FGF1 (paracrine) |
FGF1 (aFGF) | All FGFRs |
| FGF2 (bFGF) | FGFR1c, FGFR2c, FGFR3-IIIc, FGFR1b, FGFR4 | |
| FGF4 (paracrine) |
FGF4 | FGFR1c, FGFR2c, FGFR3c |
| FGF5 | FGFR1c, FGFR2 c | |
| FGF6 | FGFR2b | |
| FGF7 (paracrine) |
FGF3 | FGFR1b, FGFR2b |
| FGF7(KGF) | FGFR2b | |
| FGF10 | FGFR2b | |
| FGF22 | FGFR1b, FGFR2b | |
| FGF8 (paracrine) |
FGF8 | FGFR2c, FGFR3c, FGFR4 |
| FGF17 | FGFR2c, FGFR3c, FGFR4 | |
| FGF18 | FGFR2c, FGFR3c, FGFR4 | |
| FGF9 (paracrine) |
FGF9 | FGFR3b, FGFR3c |
| FGF16 | FGFR2c, FGFR3c, and FGFR4 | |
| FGF20 | FGFR1c | |
| FGF19 (endocrine) |
FGF15/19 | FGFR4, FGFR1c, FGFR2c, FGFR3c |
| FGF21 | FGFR1c, FGFR3c | |
| FGF23 | FGFR1c, FGFR2c, FGFR3c, FGFR4 | |
| FGF11 (Intracrine) |
FGF11 | |
| FGF12 | ||
| FGF13 | ||
| FGF14 |
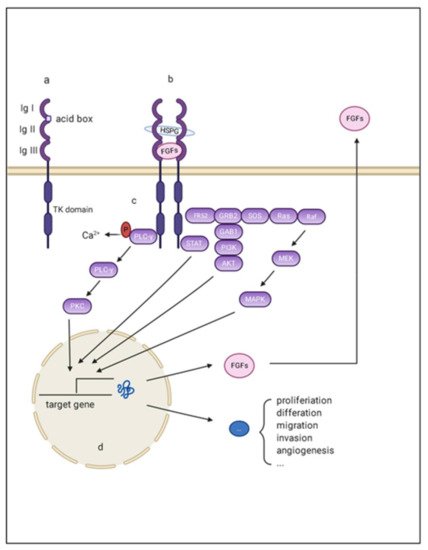
Figure 1. Schematic representation of aberrant FGF/FGFR signaling in HCC. (a) FGFR monomer structure: FGFRs are comprised of the extracellular domains linked to intracellular catalytic domains via a single pass transmembrane domain. The extra extracellular domains contains three loops (namely Ig I, Ig II, and Ig III), and an acid box with rich serine; (b) The complex composed of FGF, FGFR, and the co-factors: Ig I and the acid box is re-sponsible for signal autoinhibition, while Ig II and Ig III are essential for signal transmission through binding with FGF and the co-factors; (c) The intracellular downstream signaling of FGF/FGFR signaling: There are mainly four pathways acting as canonical downstream signaling pathways of FGF-FGFR signal: mitogen-activated protein kinase (MAPK); phosphatidylinositol 3-kinase (PI3-kinase), phospholipase Cγ (PLCγ), and signal transducer and activator of transcription (STAT); (d) The target effects of FGF/FGFR signaling: the final effects of these activating downstream pathways are transcriptionally activating a series of target genes that are responsible for multiple hallmarks of HCC.
To signal, FGFs bind to FGFR in the presence of cofactors: heparin sulfate (HS) proteoglycans (HSPGs), or Klotho protein. The affinity for different cofactors determines the endocrine, paracrine, and autocrine actions. HSPGs exist widely in the extracellular matrix. Nearly all FGFs have HS binding domains but differ in terms of affinities. On the one hand, HSPGs potentiate signaling transmission for morphogens and growth factors by functioning as accessory receptors. On the other hand, FGFs with strong association associations are tethered nearby and act in a paracrine or autocrine manner. Contrarily, FGFs with a low affinity for HSPG (FGF19, FGF21, and FGF23) enter the circulation and exert their functions through endocrine actions [16]. Without HS, those endocrine FGFs utilize klotho proteins to serve as coreceptors, which confer stability and preferential binding of endocrine FGFs to their respective FGFRs. There are two homologs of klotho protein that promote different FGF signaling pathway: β klotho (also named KLB) and α klotho (also called KL) [16]. Multiple lines of evidence demonstrate that KLB is essential for the coactivation of FGF19 and FGF21 signaling, while FGF23 tends to bind with KL to initiate the pleiotropic cellular function [17][18]. Unlike the HS in the extracellular matrix, Klotho protein has obvious tissue specificity, ultimately determining the various roles of these endocrine FGFs in different tissues. KLB is widely expressed in the liver and fat and preferentially binds with FGFR1c and FGFR4. Therefore, signals from FGFR4 and its ligand FGF19 are among the major FGF signaling pathways in HCC [19].
2.2. FGF/FGFR Downstream Signaling
To signal, secreted FGFs bind to heparin sulfate and heparin sulfate binding sites (HBS) of FGFR, forming signal-transducing dimers. Such conformational changes enable the autophosphorylation and activation of intracellular tyrosine kinases [14]. Activated FGFRs then phosphorylate docking proteins such as FGFR substrate 2 (FRS2) and FGFR substrate 3 (FRS3) [20]. These effectors can function as scaffolds and finally activate four intracellular pathways: mitogen-activated protein kinase (MAPK); phosphatidylinositol 3-kinase (PI3-kinase), phospholipase Cγ (PLCγ), and signal transducer and activator of transcription (STAT). These factors play important roles in proliferation, metastasis, angiogenesis, and agent-acquired resistance in the initiation and development of HCC (Figure 1).
3. Deregulation of FGF/FGFR in HCC
3.1. FGF2
FGF2 is expressed in HCC cells and is barely detected in nonparenchymal cells or noncancerous liver tissue [21]. By targeting the IIIc isotype of FGFR1 as its primary receptor, FGF2 is involved in the development of HCC as a potent mitogen. This molecule stimulates DNA synthesis of hepatoma cell lines and promotes tumor growth. It also enhances the synthesis of the plasminogen activator, promoting tumor cell invasion [21]. Additionally, FGF2 is a potent mitogen for endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and mural cells, participating in HCC angiogenesis [22]. FGF2 and FGF19, seem to be involved in maintaining cancer stem-like cells (CSCs) with CD44High/CD133High cell membrane markers, which play a central role in the tumor microenvironment, potentially enhancing HCC tumorigenesis, metastasis, and anticancer drug resistance [23]. Furthermore, FGF2 is reported to be involved in the immune regulation of HCC. FGF2 enhances the sensitivity of NK cells to tumor cells by upregulating the expression of membrane-bound major histocompatibility complex class I-related chain A (MICA) and suppressing the expression of human leukocyte antigen (HLA) class I, which are activating molecule and inhibitory molecules of NK cells, respectively [24]. From this point of view, FGF2 may partly contribute to the elimination of innate immunity in HCC cells. GAL-F2 is a specific monoclonal antibody for FGF2. This molecular inhibits the proliferation and migration of HCC cell lines and blocks angiogenic signals in the mouse model, corroborating the role of FGF2 in tumor growth and vascularization [25].
3.2. FGF8 Subfamily
Although widely expressed during embryonic development, the expression of FGF8 subfamilies is mainly restricted to hormonal cancers such as prostate cancer and breast cancer during adulthood [26]. However, the abnormal signaling of FGF8 subfamilies (FGF8, FGF17 and FGF18) have also been detected in many other cancers [8][26][27]. By a paracrine and autocrine mechanism, FGF8, FGF17, and FGF18 participate in the development of HCC. In vitro, FGF8 promotes the proliferation of HCC cell lines. Moreover, FGF8 increases the expression of EGFR through transcriptionally upregulating Yes-associated protein 1 (YAP1), which contributes to resistance to EGFR inhibitors [28]. FGF17 is a potent mitogen for prostate cancer. This molecule can be induced by FGF8, indicating its potential mediating role in FGF8 function [29]. FGF18 impaires apoptosis while enhancing cell proliferation, motility, and invasion in HCC [30]. Knockout of FGF18 dramatically reduces the malignant phenotype of cells [31]. According to research conducted by Gauglhofer et al. [26], the levels of at least one FGF8 subfamily member and/or one FGFR are upregulated in 82% of HCC cases. Additionally, the co-upregulation of the levels of at least one FGF and one FGFR is detected in approximately one-third of these tumor. The researchers also noted that the Wnt pathway and hypoxia-inducible transcription factors might be two possible mechanisms that regulated FGF8, FGF17 and FGF18 overexpression in HCC [26]. In addition to contributing to the tumorigenic characteristics of tumor cells, FGF18 subfamilies are also involved in the angiogenesis of HCC, which will be discussed in the subsequent chapters.
3.3. FGF9
FGF9 expression is often co-upregulated with FGFR3 IIIb/IIIc expression in HCC patients. Furthermore, FGF9 is the most dominant ligand for FGFR3 IIIb/IIIc in HCC [32]. FGF9 is secreted mainly by HSCs in normal and cirrhotic livers and acts by the paracrine mechanism to stimulate tumor cells. However, this molecule has an autocrine effect in HCC since tumor cells are the primary source at this stage. This result is inconsistent with other research, which noted out that activated HSCs/myofibroblasts are the primary sources of FGF9, while no FGF9 expression was detected in either HCC cells and hepatocytes [33]. However, both findings emphasize the tumorigenic role of FGF9 in HCC. FGF9 exerts its tumorigenic role by specifically binding to FGFR3, facilitating the proliferation and migration of HCC cell lines and promoting new blood and lymphatic capillary formation [32]. Moreover, FGF9 is involved in sorafenib resistance, indicating that FGF9 may be a promising target of HCC. Pan-inhibitors of FGFR or siRNA targeting FGFR3 could block the oncogenic properties of FGF9 in HCC [33].
3.4. FGF19 Subfamily
This family is closely associated with multiple metabolic regulatory processes, including insulin resistance, fatty acid oxidation, bile acid, triglycerides, and glycogen [6]. Many of these metabolic processes are found in the liver and have close relationships with liver physiology and pathology status. FGF19, FGF21, and FGF23 can facilitate HCC in both metabolism-dependent and metabolism-independent pathways. This chapter will discuss the metabolism-independent roles, and the metabolic effects in HCC development will be discussed in the following section.
In hepatocytes, FGF19 mainly targets FGFR4 as its receptor, with KLB stabilizing their integration and interaction. FGF19 is primarily expressed and secreted from ileum villus epithelial cells and gallbladder epithelial cells in adults. FGF19 can also be secreted by cells from pathological liver tissue, such as cholestatic noncirrhotic and cirrhotic livers and livers from individuals with alcoholic hepatitis and HCC [34]. Additionally, FGF19, FGFR4, and KLB appear to increase with hepatic pathology, from steatosis to steatohepatitis, cirrhosis, and finally HCC [35]. Transgenic mice with ectopically expressed FGF19 exhibited preneoplastic changes, including constitutive hepatocellular proliferation and AFP expression. At 10–12 months, an average of 53% of the mice had developed locally invasive HCCs. In p53−/− mice, all FGF19 transfected mice died within the 100-day observation period, while none of those in the control groups died [36]. These results support the direct effect of FGF19 on HCC initiation [37]. With the coaction of KLB, FGFR4 initiates a growing number of intracellular signaling pathways to target tumor cells, promoting hepatoma cell proliferation and migration and inhibiting tumor cells apoptosis. FGF19 mediates cell escape from death by increasing the expression and phosphorylation of IL-6-induced STAT3, which is known to lead to compensatory proliferation in tumor cells [38][39]. Another newly discovered target gene of FGF19 in HCC is SOX18. SOX18 is an oncogene promoting the proliferation and metastasis of tumor cells in many cancers. FGF19/FGFR4 upregulates the expression of SOX18 through p-FRS2/p-GSK3β/β-catenin signaling. Interestingly, SOX18 is also a ligand for FGFR4, forming positive feedback among SOX18, FGF19, and FGFR4 in HCC development. BLU9931, which is a selective FGFR4 inhibitor, significantly inhibits the growth of SOX18-induced HCC metastasis [40].
However, FGF19 also exerts protective effects on the liver. Mitogenic FGF19 deficiency delays liver regrowth and impairs hepatocyte regeneration after chemical liver injury or partial hepatectomy in a mouse model. Similarly, the knockout of FGFR4 or siRNA application increases the susceptibility of the liver to CCL4 exposure. This phenomenon mechanism can be partly attributed to bile acid accumulation resulting from FGF19 deficiency. Additionally, the proliferative signals provided by FGF15 are also indispensable for the regeneration since a cholate-supplemented diet cannot compensate for the growth impairment in FGF15-null mice [41]. NF2/Merlin might control the shunting of pro-oncogenic and antioncogenic signaling of FGFR4. NF2/Merlin is an upstream regulator of the Hippo pathway and is activated by FGFR4 signaling to maintain various organ sizes. NF2/Merlin might act as a switch in FGFR4 signaling by activating ERK and attenuating Mst1/2-mediated signaling [42].
Unlike other FGFs in HCC, FGF21 expression is usually decreased in HCC and is believed to protect the liver. Multiple lines of evidence have shown that FGF21 maintains metabolic homeostasis and contributes to antifibrotic processes during the development of HCC [43][44]. FGF21 is reported to relieve acute or chronic inflammatory diseases by inhibiting the production of IL-17A, which has recently been proven to be associated with human hepatitis, fatty livers, and viral hepatitis-associated HCC [45]. Both rhFGF21 administration and blockage of IL-17A benefited the liver in terms of arresting progressive liver diseases [44][45].
3.5. Other FGFs in HCC
FGF5 has been discussed in many other cancers and has multiple roles in cancer development [46][47][48]. FGFR1 IIIc and FGFR2 IIIc are considered the preferential receptors for FGF5. In pancreatic cancer, FGF5 promotes pancreatic cancer cells growth through its binding to FGFR1 IIIc [47]. Fang et al. [49] noted that FGF5, as a downstream molecule of miR-188-5p, is involved in the proliferation, colony formation, cell migration, and invasion of SMMC7721 cells, promoting carcinogenesis in HCC by activating H-Ras—p-ERK signaling. However, no research has been conducted to identify the exact receptor of FGF5 in HCC and its biological functions in vivo. FGF7 has been reported to participate in the nucleotide excision repair (NER) pathway by upregulating ERCC1 expression via the FGFR2-ERK pathway [50]. ERCC1 has been identified as a critical rate-limiting enzyme during the NER process. The overexpression of ERCC1 reflects the higher activity of NER related to HCC resistance to platinum drugs [51].
While some FGFs may have an oncogenic role in other organs, their roles in HCC remain to be clarified. These FGFs include FGF6, FGF10, FGF20, and FGF22. FGF6 is strongly overexpressed in prostate cancer tissues compared with normal prostate tissues, stimulating the transformed of prostatic epithelial cells [52]. However, FGF6 poorly expressed in normal liver tissues and HCC, and it accumulates almost exclusively in the myogenic lineage [53]. FGF10 is involved in multiorgan development, and its knockout may cause severe dysmorphia [54]. Otherwise, FGF10 is thought to act as a mediator of androgen action, thus contributing to prostate cancer pathogenesis by facilitating epithelial proliferation [55]. FGF22 in the brain is reported to be associated with depression. To date, no research has been performed to illustrate the role of these FGFs in HCC.
3.6. FGFRs in HCC
FGFR3 and FGFR4 are the major FGFRs overexpressed in HCC, while upregulation of FGFR1 and FGFR2 expressing are rarely observed [56][57]. FGFR3 expression has been reported to be upregulated in 17 of 32 HCC cases. In five cases displaying upregulation of IIIb expression, eight cases showed upregulation of IIIc expression, and four cases showed both. The ligands of FGFR3 are different according to the variants. The ligands of the FGFR3-IIIb variants are mainly FGF1 and FGF9. In comparison, the FGFR3-IIIc variants have additional binding sites for FGF2, FGF4, FGF8, FGF17, FGF18, FGF19, FGF21, and FGF23. Although the two isoforms provide different FGFs docking sites and transmit different downstream signals, upregulation of the expression of both enhances hepatoma cells malignant phenotypes [57]. FGFR4 shows the highest expression in the liver compared with other major organs. Additionally, hepatocyte is the only cell type where FGFR4 is more dominant than all other FGFRs [58]. In HCC, FGFR4 expressions is upregulated in nearly half of HCC cases, along with a major increase in different FGF ligands, such as the FGF2 and FGF8 subfamilies [59]. Although all of these ligands can bind with FGFR4, signals from FGF19 seem to be exerted preferentially and cannot be replaced by any other ligands [59].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13061360
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6.
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428.
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604.
- Roh, M.S. Recent progress in the treatment of hepatocellular carcinoma. Curr. Opin. Oncol. 1990, 2, 725–730.
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129.
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181.
- Sharpe, R.; Pearson, A.; Herrera-Abreu, M.T.; Johnson, D.; Mackay, A.; Welti, J.C.; Natrajan, R.; Reynolds, A.R.; Reis-Filho, J.S.; Ashworth, A.; et al. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 2011, 17, 5275–5286.
- Jomrich, G.; Hudec, X.; Harpain, F.; Winkler, D.; Timelthaler, G.; Mohr, T.; Marian, B.; Schoppmann, S.F. Expression of FGF8, FGF18, and FGFR4 in Gastroesophageal Adenocarcinomas. Cells 2019, 8, 1092.
- Stehbens, S.J.; Ju, R.J.; Adams, M.N.; Perry, S.R.; Haass, N.K.; Bryant, D.M.; Pollock, P.M. FGFR2-activating mutations disrupt cell polarity to potentiate migration and invasion in endometrial cancer cell models. J. Cell Sci. 2018, 131, jcs213678.
- Szybowska, P.; Kostas, M.; Wesche, J.; Wiedlocha, A.; Haugsten, E.M. Cancer Mutations in FGFR2 Prevent a Negative Feedback Loop Mediated by the ERK1/2 Pathway. Cells 2019, 8, 518.
- Chae, Y.K.; Hong, F.; Vaklavas, C.; Cheng, H.H.; Hammerman, P.; Mitchell, E.P.; Zwiebel, J.A.; Ivy, S.P.; Gray, R.J.; Li, S.; et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: Results from the NCI-MATCH Trial (EAY131) subprotocol W. J. Clin. Oncol. 2020, 38, 2407–2417.
- Olsen, S.K.; Garbi, M.; Zampieri, N.; Eliseenkova, A.V.; Ornitz, D.M.; Goldfarb, M.; Mohammadi, M. Fibroblast Growth Factor (FGF) Homologous Factors Share Structural but Not Functional Homology with FGFs. J. Biol. Chem. 2003, 278, 34226–34236.
- Ornitz, D.M.; Itoh, N. Protein family review: Fibroblast growth factors. Genome Biol. 2001, 2, 1–12.
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 2012, 20, 77–88.
- Holzmann, K.; Grunt, T.; Heinzle, C.; Sampl, S.; Steinhoff, H.; Reichmann, N.; Kleiter, M.; Hauck, M.; Marian, B. Alternative splicing of fibroblast growth factor receptor IgIII loops in cancer. J. Nucleic Acids 2012, 2012.
- Kuro-o, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44.
- Kuzina, E.S.; Ung, P.M.U.; Mohanty, J.; Tome, F.; Choi, J.; Pardon, E.; Steyaert, J.; Lax, I.; Schlessinger, A.; Schlessinger, J.; et al. Structures of ligand-occupied β-Klotho complexes reveal a molecular mechanism underlying endocrine FGF specificity and activity. Proc. Natl. Acad. Sci. USA 2019, 116, 7819–7824.
- Hu, M.C.; Shiizaki, K.; Kuro-O, M.; Moe, O.W. Fibroblast growth factor 23 and klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Physiol. 2013, 75, 503–533.
- Alvarez-Sola, G.; Uriarte, I.; Ujue Latasa, M.; Urtasun, R.; Bárcena-Varela, M.; Elizalde, M.; Jiménez, M.; Rodriguez-Ortigosa, C.M.; Corrales, F.J.; Fernández-Barrena, M.G.; et al. Fibroblast Growth Factor 15/19 in Hepatocarcinogenesis. Dig. Dis. 2017, 35, 158–165.
- Ong, S.H.; Hadari, Y.R.; Gotoh, N.; Guy, G.R.; Schlessinger, J.; Lax, I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment. Proc. Natl. Acad. Sci. USA 2001, 98, 6074–6079.
- Kin, M.; Sata, M.; Ueno, T.; Torimura, T.; Inuzuka, S.; Tsuji, R.; Sujaku, K.; Sakamoto, M.; Sugawara, H.; Tamaki, S.; et al. Basic fibroblast growth factor regulates proliferation and motility of human hepatoma cells by an autocrine mechanism. J. Hepatol. 1997, 27, 677–687.
- Nissen, L.J.; Cao, R.; Hedlund, E.M.; Wang, Z.; Zhao, X.; Wetterskog, D.; Funa, K.; Bråkenhielm, E.; Cao, Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J. Clin. Investig. 2007, 117, 2766–2777.
- Shigesawa, T.; Maehara, O.; Suda, G.; Natsuizaka, M.; Kimura, M.; Shimazaki, T.; Yamamoto, K.; Yamada, R.; Kitagataya, T.; Nakamura, A.; et al. Lenvatinib suppresses cancer stem-like cells in HCC by inhibiting FGFR1–3 signaling, but not FGFR4 signaling. Carcinogenesis 2021, 42, 58–69.
- Tsunematsu, H.; Tatsumi, T.; Kohga, K.; Yamamoto, M.; Aketa, H.; Miyagi, T.; Hosui, A.; Hiramatsu, N.; Kanto, T.; Hayashi, N.; et al. Fibroblast growth factor-2 enhances NK sensitivity of hepatocellular carcinoma cells. Int. J. Cancer 2012, 130, 356–364.
- Wang, L.; Park, H.; Chhim, S.; Ding, Y.; Jiang, W.; Queen, C.; Kim, J.K. A Novel Monoclonal Antibody to Fibroblast Growth Factor 2 Effectively Inhibits Growth of Hepatocellular Carcinoma Xenografts. Bone 2014, 23, 1–7.
- Gauglhofer, C.; Sagmeister, S.; Schrottmaier, W.; Fischer, C.; Rodgarkia-Dara, C.; Mohr, T.; Stättner, S.; Bichler, C.; Kandioler, D.; Wrba, F.; et al. Upregulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology 2011, 53, 854–864.
- Harpain, F.; Ahmed, M.A.; Hudec, X.; Timelthaler, G.; Jomrich, G.; Müllauer, L.; Selzer, E.; Dörr, W.; Bergmann, M.; Holzmann, K.; et al. FGF8 induces therapy resistance in neoadjuvantly radiated rectal cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 77–86.
- Pei, Y.; Sun, X.; Guo, X.; Yin, H.; Wang, L.; Tian, F.; Jing, H.; Liang, X.; Xu, J.; Shi, P. FGF8 promotes cell proliferation and resistance to EGFR inhibitors via upregulation of EGFR in human hepatocellular carcinoma cells. Oncol. Rep. 2017, 38, 2205–2210.
- Heer, R.; Douglas, D.; Mathers, M.E.; Robson, C.N.; Leung, H.Y. Fibroblast growth factor 17 is over-expressed in human prostate cancer. J. Pathol. 2004, 204, 578–586.
- Yang, L.; Yin, D.; Wang, Y.; Cao, L. Inhibition of the growth of hepatocellular carcinoma cells through fibroblast growth factor 18 suppressed by miR-139. Oncol. Rep. 2017, 38, 2565–2571.
- Zhang, J.; Zhou, Y.; Huang, T.; Wu, F.; Pan, Y.; Dong, Y.; Wang, Y.; Chan, A.K.Y.; Liu, L.; Kwan, J.S.H.; et al. FGF18, a prominent player in FGF signaling, promotes gastric tumorigenesis through autocrine manner and is negatively regulated by miR-590-5p. Oncogene 2019, 38, 33–46.
- Paur, J.; Valler, M.; Sienel, R.; Taxauer, K.; Holzmann, K.; Marian, B.; Unterberger, A.; Mohr, T.; Berger, W.; Gvozdenovich, A.; et al. Interaction of FGF9 with FGFR3-IIIb/IIIc, a putative driver of growth and aggressive behavior of hepatocellular carcinoma. Liver Int. 2020, 40, 2279–2290.
- Seitz, T.; Freese, K.; Dietrich, P.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Fibroblast Growth Factor 9 is expressed by activated hepatic stellate cells and promotes progression of hepatocellular carcinoma. Sci. Rep. 2020, 10, 1–9.
- Wang, Y.; Gunewardena, S.; Li, F.; Matye, D.J.; Chen, C.; Chao, X.; Jung, T.; Zhang, Y.; Czerwiński, M.; Ni, H.M.; et al. An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis. Nat. Commun. 2020, 11, 1–16.
- Li, Y.; Zhang, W.; Doughtie, A.; Cui, G.; Li, X.; Pandit, H.; Yang, Y.; Li, S.; Martin, R. Upregulation of fibroblast growth factor 19 and its receptor associates with progression from fatty liver to hepatocellular carcinoma. Oncotarget 2016, 7, 52329–52339.
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a Therapeutic Strategy Targeting Amplified FGF19 in Liver Cancer by Oncogenomic Screening. Cancer Cell 2011, 19, 347–358.
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.-P.; et al. A Mouse Model of Hepatocellular Carcinoma. Am. J. Pathol. 2002, 160, 2295–2307.
- Massafra, V.; Milona, A.; Vos, H.R.; Burgering, B.M.T.; Van Mil, S.W.C. Quantitative liver proteomics identifies FGF19 targets that couple metabolism and proliferation. PLoS ONE 2017, 12, 1–18.
- Li, T.; Zhang, G.; Wang, L.; Li, S.; Xu, X.; Gao, Y. Defects in mTORC1 Network and mTORC1-STAT3 Pathway Crosstalk Contributes to Non-inflammatory Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2020, 8, 1–14.
- Chen, J.; Du, F.; Dang, Y.; Li, X.; Qian, M.; Feng, W.; Qiao, C.; Fan, D.; Nie, Y.; Wu, K.; et al. Fibroblast Growth Factor 19–Mediated Upregulation of SYR-Related High-Mobility Group Box 18 Promotes Hepatocellular Carcinoma Metastasis by Transactivating Fibroblast Growth Factor Receptor 4 and Fms-Related Tyrosine Kinase 4. Hepatology 2020, 71, 1712–1731.
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Jimenez, M.; Barcena-Varela, M.; Santamaría, E.; Urtasun, R.; Rodriguez-Ortigosa, C.; Prieto, J.; Berraondo, P.; et al. Bile acids, FGF15/19 and liver regeneration: From mechanisms to clinical applications. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1326–1334.
- Ji, S.; Liu, Q.; Zhang, S.; Chen, Q.; Wang, C.; Zhang, W.; Xiao, C.; Li, Y.; Nian, C.; Li, J.; et al. FGF15 Activates Hippo Signaling to Suppress Bile Acid Metabolism and Liver Tumorigenesis. Dev. Cell 2019, 48, 460–474.e9.
- Liu, W.Y.; Huang, S.; Shi, K.Q.; Zhao, C.C.; Chen, L.L.; Braddock, M.; Chen, Y.P.; Feng, W.K.; Zheng, M.H. The role of fibroblast growth factor 21 in the pathogenesis of liver disease: A novel predictor and therapeutic target. Expert Opin. Ther. Targets 2014, 18, 1305–1313.
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936.
- Gomes, A.L.; Teijeiro, A.; Burén, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175.
- Allerstorfer, S.; Sonvilla, G.; Fischer, H.; Gauglhofer, C.; Setinek, U.; Marosi, C.; Buchroithner, J.; Pichler, J.; Silye, R.; Mohr, T.; et al. UKPMC Funders Group autocrine and paracrine activities. Nature 2010, 27, 4180–4190.
- Kornmann, M.; Ishiwata, T.; Beger, H.G.; Korc, M. Fibroblast growth factor-5 stimulates mitogenic signaling and is overexpressed in human pancreatic cancer: Evidence for autocrine and paracrine actions. Oncogene 1997, 15, 1417–1424.
- Zhou, Y.; Yu, Q.; Chu, Y.; Zhu, X.; Deng, J.; Liu, Q.; Wang, Q. Downregulation of fibroblast growth factor 5 inhibits cell growth and invasion of human nonsmall-cell lung cancer cells. J. Cell. Biochem. 2019, 120, 8238–8246.
- Fang, F.; Chang, R.M.; Yu, L.; Lei, X.; Xiao, S.; Yang, H.; Yang, L.Y. MicroRNA-188-5p suppresses tumor cell proliferation and metastasis by directly targeting FGF5 in hepatocellular carcinoma. J. Hepatol. 2015, 63, 874–885.
- Chen, G.; Qiu, H.; Ke, S.; Hu, S.; Yu, S.; Zou, S. The fibroblast growth factor receptor 2-mediated extracellular signal-regulated kinase 1/2 signaling pathway plays is important in regulating excision repair cross-complementary gene 1 expression in hepatocellular carcinoma. Biomed. Rep. 2013, 1, 604–608.
- Fautrel, A.; Andrieux, L.; Musso, O.; Boudjema, K.; Guillouzo, A.; Langouët, S. Overexpression of the two nucleotide excision repair genes ERCC1 and XPC in human hepatocellular carcinoma. J. Hepatol. 2005, 43, 288–293.
- Ropiquet, F.; Giri, D.; Kwabi-addo, B.; Mansukhani, A.; Ittmann, M. Increased expression of fibroblast growth factor 6 in human prostatic intraepithelial neoplasia and prostate cancer. Cancer Res. 2000, 60, 4245–4250.
- Armand, A.S.; Laziz, I.; Chanoine, C. FGF6 in myogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 773–778.
- Komiya, A.; Watanabe, A.; Yasuda, K.; Fuse, H. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Nippon Rinsho. Jpn. J. Clin. Med. 2010, 68 (Suppl. S7), 505–510.
- Li, Q.; Ingram, L.; Kim, S.; Beharry, Z.; Cooper, J.A.; Cai, H. Paracrine Fibroblast Growth Factor Initiates Oncogenic Synergy with Epithelial FGFR/Src Transformation in Prostate Tumor Progression. Neoplasia 2018, 20, 233–243.
- Qiu, W.H.; Zhou, B.S.; Chu, P.G.; Chen, W.G.; Chung, C.; Shih, J.; Hwu, P.; Yeh, C.; Lopez, R.; Yen, Y. Over-expression of fibroblast growth factor receptor 3 in human hepatocellular carcinoma. World J. Gastroenterol. 2005, 11, 5266.
- Paur, J.; Nika, L.; Maier, C.; Moscu-Gregor, A.; Kostka, J.; Huber, D.; Mohr, T.; Heffeter, P.; Schrottmaier, W.C.; Kappel, S.; et al. Fibroblast growth factor receptor 3 isoforms: Novel therapeutic targets for hepatocellular carcinoma? Hepatology 2015, 62, 1767–1778.
- Ho, H.K.; Pok, S.; Streit, S.; Ruhe, J.E.; Hart, S.; Lim, K.S.; Loo, H.L.; Aung, M.O.; Lim, S.G.; Ullrich, A. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J. Hepatol. 2009, 50, 118–127.
- Gauglhofer, C.; Paur, J.; Schrottmaier, W.C.; Wingelhofer, B.; Huber, D.; Naegelen, I.; Pirker, C.; Mohr, T.; Heinzle, C.; Holzmann, K.; et al. Fibroblast growth factor receptor 4: A putative key driver for the aggressive phenotype of hepatocellular carcinoma. Carcinogenesis 2014, 35, 2331–2338.
This entry is offline, you can click here to edit this entry!